Diseases Flashcards
Causes for iron deficiency?
BLOOD LOSS #1
iron-poor diet
malabsorption
20% pre-menopausal women, 50% preggers
What is hemochromatosis?
genetic disorder, autosomal recessive
caused by mutation in HFE gene mostly
patients absorb excessive iron
iron deposits in liver, heart, pancreas, pituitary gland, other tissues
since liver affected, risk of cirrhosis and hepatocellular carcinoma
since pancreas affected - damage to islet cells -> DM in 2/3 of patients
increase in skin pigmentation as a result of increased melanin and increased iron deposition within the skin
cardiac deposition -> cardiac damage, CHF, arrhythmias
pituitary land iron -> drop in production of gonadotropins (sex hormones) -> testicular atrophy, etc
arthritis common - up to 1/2 of patients
takes years to develop serious disease - usually presents in mid 30ies-40ies
-> patients with hemochromatosis do not make enough hepscidin
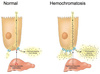
Acquired hemochromatosis?
iron overload can be acquired
first deposited into RE cells of the liver, spleen, bone marrow (R - connective tissue cells with reticulocytes - special form of collagen; RE cells = macrophages that break down RBCs)
as RE cells saturate, iron deposited in liver, pancreas, heart and other organs -> resembles hereditary
can be complication of anemias with ineffective erythropoiesis, such as beta-thalassemia - iron absorption increased and multiple blood transfusions add even more iron
alcoholics with liver disease can develop increased iron stores, since hepcidin made in the liver
increased iron consumption by itself does not cause hemochromatosis - body regulates absorption
Causes of B12 deficiency?
- poor nutrition (vegans)
- pernicious anemia (ex. pariental cells damaged - no IF = intrinsic factor)
- total or partial gastrectomy (since need IF from stomach to transport B12 to intestine for absorption)
- intestinal disease (need intestinal factors to transport and absorb)
Causes of folate deficiency?
- poor nutrition (elderly, powerty, alcoholics)
- increased utilization (pregnancy, lactation, malignancy - need for DNA synthesis, inflammation, etc)
- intestinal disease (link to B12)
- drug induced
What is megaloblastic anemia (do not confuse with macrocytic anemia)?
- B12 and folate deficiency most common causes of megaloblastic anemia, since DNA synthesis is defective
- RBCs (erythroblasts) in bone marrow show delayed maturation of nucleus relative to cytoplasm - cells bigger b/c cytoplasm grows more before nucleus can mature
- circulating RBCs are large (macrocytic) with marked variation in size and shape
- megaloblastic anemia is one of the types of microcytic anemia

Role of folate and B12 in blood?
B12 - cofactor for two biochem reactions
- homocysteine-> methione which generates tetrahydrofolate (THF) from methyl THF. need tetrahydrofolate because that is the active form of folate inside cells
- methylmalonyl coA -> Succinyl CoA (part of citric acid/Krebs - in the middle of the cycle)
Folate
cofactor in numerous biochem reactions
required to synthesize a precursor of DNA
both important for RBC maturation -> not enough -> DNA sythesis for RBC slows down, cytoplasm increases in relation to nucleus -> larger cells = macrocytic anemia
List mechanisms for anemia (3)?
* blood loss
* decreased production
* increased destruction
What is reticulocyte count?
- reticulocyte count - marrow’s ability to produce young RBCs in response to turnover reticulocyte - immature RBC, 1% or so normal
- increased reticulocyte count - adequate production of young RBCs to satisfy decreased RBC destruction (hemolysis, recovery post hemorrhage, therapy -ex. Fe-deficiency anemia and you give them iron)
- decreased reticulocyte count - inadequate stores (Fe, B12, folate - slow RBC production) or to marrow abnormality (decreased ability to produce RBCs)
- interpret with Hb
Outline approach to diagnosing anemia based on RBC size?
Microcytic = decreased MCV ) ~ <82
Normocytic = normal MCV (mean corpuscular volume) 82-98
Macrocytic = increased MCV -100+
List differentials for microcytic anemia?
- small TAILS
- Thalassemia (missing one of the Hb chains)
- Anemia of chronic disease (b/c reduced iron release from RE macrophages - less iron available to be used for RBC synthesis) = also in malignancy and infection
- Iron deficiency (b/c not enough building blocks for Hb)
- Lead poisoning
- Sideroblastic anemia
Discuss pathogenesis of iron deficiency anemia?
- iron is part of Hb and rate-limiting for erythropoiesis
Causes (decreased intake, decreased absorption = increased elimination)
- # 1 - GI (men) or uterine (women) bleeding
- bleeding from GI tract
- menorrhagia (heavy or prolonged menstrual period)
- pregnancy
- malabsorption (celiac, gastritis, GI malignancy, hookworm)
- malnutrition
- bleeding from GU
Common symptoms of anemia?
- SOB
- fatigue
- dizziness
- weakness
- pallor
- palpitations - heart starts beating faster to maintain adequate O2 supply
- tinnitus (ringing in ears)
- pica (appetitne for non-food substances, like ice or dirt)
- weak hair, nail flattening and concaven nails = koilonychia
- conjunctival pallor
Investigations for anemia?
- # 1 CBC and peripheral blood smear
- look for cause - consider endoscopy or colonoscopy if change in bowel habits or new indigestion
- fecal occult blood test is useful to exclude GI bleed
- can test urine for hematuria
- chest X-ray to exclude very rare pulmonary haemosiderosis
- sometimes no cause found (20%)
*
Results of investigations?
- low ferritin is diagnostic of iron deficiency! (ferritin = storage form of iron, iron wrapped in proteins)
- ferritin is also an acute phrase reactant iand is elevated in inflammatory condtions and liver disease
- peripheral blood film shows hypochromic microcytosis - small RBCs with large white centre (hypochromic), can also have target cells
- increased TIBC (iron wants to bind to transferrin more to be transported to tissues)
- decreased serum ferum (iron bound to transferrin low, b/c low iron)
- decreased iron saturation - less iron bound to transferrin b/c less iron in general

Treatment for anemia?
- treat underlying cause
- supplement iron
- oral - tablets/syrup - ferrous sulphate or ferrous gluconate (ferrous = Fe2+)
- IV if cannot tolerate or absorb iron (iron sucrose or dextran)
- monitor response - want increased reticulocyte count, increased Hb, will need to be on supplements for 4-6 months
Pathogenesis of alpha thalassemia?
Defect in alpha globulin gene - decreased synthesis
4 alleles total for alpha globulin
if all 4 missing - no alpha globulin - incompatible with life - Hb Barts (gamma 4) - hydrops fetalis
3 allele deletion - HbH disease - very little alpha globulin - excess beta globin forms beta 4 Hb (HbH) - decreased MCV (mean cell volume), decreased Hb, splenomegaly (breakdown of malfunctioning alpha globulins)
1-2 allele deletion - virtually asymptomatic
alpha for A = Asia = South East Asia (SEA) and Africa
HbA 97% of adult hemoglobin
HbA2 - 3% of adult hemoglobin
both contain two alphas
Pathogenesis for beta thalassemia?
Point mutation - decreased beta blobin synthesis
prevalent in mediterranean populations
beta -thalassemia minor (heterozygote) - less beta chains, usually asymptomatic
diagnosis confirmed by increased HbA2 (which has 2 alphas and 2 gammas (no betas) on electrophoresis
beta-thalassemia major (homozygote) - absent beta -> severe anemia, requiring blood transfusions -> can lead to secondary hemochromatosis ; here autosomal recessive
marrow expansion
increased HbF (2 alpha 2 gamma) - HbF naturally exists until 6 months, so infant only symptomatic after 6 months, when HbA takes over -> severe anemia and jaundice, then stunted growth and development, hepatosplenomegaly (extramedullary hematopoiesis - outside of bone marrow), expanded bone marrow - b/c of increased RBC synthesis
HbS (sickle) or beta thalassemia heterozygote - mild to moderate sickle sickle cell disease depending on amount of beta globulin
- marrow expands in an effort to produce more RBCs (hemoglobin not as effective in transporting O2, so need more RBCs) -> bone enlargements (ex. ‘hair-on-end’ fluffy appearance on Xrays of skull) -> if untreated bone fractures, ulcers, etc
Testing for alpha thalassemia and treatment?
Testing:
peripheral blood film - screen for HbH inclusion bodies with special stain
Hb electorphoresis not diagnostic for apha-thalasssemia - 4 copies of alpha
DNA analysis is the only way to confirm
**Treatment: **
depends on degree: if 1 or 2 defective alleles - no treatment required
if 3 (HbH disease) - transfuse
if 4 (HbBarts) - intrauterine transfusion (incompatible with life - not born otherwise)
Testing and treatment for beta thalassemia?
Investigations:
CBC - low Hb
increased # RBCs - body tries to compensate for low Hb by making more RBCs -but RBCs smaller - missing Hb particles - microcytic anemia - also leads to bigger bones on Xrays (hematopoiesis is in bone marrow, breakdown in spleen - splenomegaly)
Hb electrophoresis: HbA really low (b/c beta parts missing), HbA2 high (b/c does not contain beta), HbF high (even though should be at 0 after 6 months
Treatment:
lifelong transfusions to stop endogenous production of Hb
iron chelation because increased transfusions lead to hemochromatosis
splenectomy - now less frequent
common in mediterranean patients
Describe pathogenesis, tests and treatment for lead poisoning?
**Pathogenesis: **
lead inhibits enzymes involved in heme synthesis and increases RBC protoporphyrin (empty porphyrin without Fe), also lead inhibits rRNA degradation, which causes RBCs to have basophilic stippling (purple basophilic dots)
high risk in old houses with chipped paint
Test:
blood lead levels - consider occupational history
abdo pain, constipatino, irritability, lack of concentration
Treat: chelation therapy
LEAD
Lead Lines on gingivae (Burton lines) and on long bones on x-rays
Encephalopathy and erythrocyte basophilic stippling (purple dots)
Abdominal colic and sideroblastic Anemia
Drops - wrist and foot drop. Dimercaprol and eDta to chelate, Succimer in kids - “sucks to be a kid who eats lead
Discuss pathogenesis of anemia of chronic disease?
Anemia of inflammatory, infective and malignant disesases (malignancy, RA, connective tissue disorders, chronic infections or extensive trauma)
- inflammation -> IL6 released -> stimulates hepcidin release from liver -> hepcidin inhibits ferroportin (channel that pumps iron into body from enterocyte) -> reduced plasma levels
- macrophage trapping iron -> reduced plasma levels
- marrow unresponsive to normla or slightly elevated EPO - reduced RBC
rarely severe (Hb usually >90 vs 130 N in males, 120N in females)
very common - probably #2 after iron deficiency
inflammatory cytokines, such as IL-1 and IL-6 -> esp IL-6 can affect hepcidin (released from liver) directly, hepcidin binds to ferroportin and blocks absorption of iron from GI -> decreased plasma iron concentration
thought that maybe hits reduces Hb-O2 carrying capacity - limits O2 to microorganisms to help fight infection
anemia of chronic disease -> decreased iron levels - > decreased RBC production (also in part due to blunted response to erythropoietin and reduced red cell survival)
Tests for anemia of chronic disease?
- elevation in acute phase reactants (CRP, fibrinogen, ferritin)
- serum iron and TIBC low b/c lower circulating iron, but % saturation normal, ferritin normal (iron stores not affected)
- Hb slightly down
- serum iron concentration and transferrin concentration are reduced (less iron)
- serum ferritin level is normal or high - serum ferritin is acute phase reactant and increases in inflammation, malignancy and infection
- normocytic (mild anemia of chronic disease) or sometimes microcytic (moderate anemia of chronic disease)
Management of anemia of chronic disease?
- manage underlying disorder
- usually condition non-severe and non-progressive; treat only if Hb too low
- IV iron if PO not working
- if cannot compensate, consider blood transfusion, can give erythropoietin if chronic renal failure
Summarize blood findings in microcytic anemias?

What is sideroblastic anemia?
Sideroblasts are erythrocytes with iron-containing (basophilic) granules in the cytoplasm
“normal” iron-containing granules are small and randomly spread in the cytoplasm in healthy individuals.
“ring” : iron deposits in mitochondria, forming a ring around the nucleus - large granules in sideroblastic anemia - this is from bone marrow b/c peripheral blood would not show nucleus or ribosomes
“ring sideroblasts are found exclusively in pathological conditions, and should not be confused with ferritin sideroblasts, which are present in normal bone marrow. “
due to defect in heme synthesis - hereditary, X-linked or acquired (ex. myelodysplastic syndrome) or drug-related

Investigations and treatment of sideroblastic anemia?
Investigations:
- increased serum Fe2+, normal TIBC, increased ferritin (increased iron, which is what produces basophilic stippling)
- ringed sideroblasts on peripheral blood smear (mitochondria with lots of iron around nucleus)
- RBCs are hypochromic, basophilic stripping
- can be micro, normo or macrocytic
Treatment:
depends on etiology
Pathophysiology of sickle cell anemia?
part of hemoglobinopathies, primarily affects Afro-Carribean population (thought to be protective against malaria, since RBC changes shape and parasite cannot inhabit it)
betaSbeta S - serious disorder
betaSbetaThalassemia - serious, similar
betaSbetaNormal - sickle trait
betaS - one of the amino acids in Hb sequence replaced - in deoxygenated state HbS changes shape from flat tetramer to tetramers that pile on top of each other making a long polymer. cell looses its ability to restore shape and stays sickle shaped due to membrane damage -> sequestration of RBCs in microvasculature -> extreme pain and ischemia and increased hemolysis due to sequestration in RE network (like spleen)
due to this hemolysis, make sure patient on folate to prevent further drop in RBCs
Symptoms and complications of sickle cell anemia?
- haemolytic anemia
RBCs do not retain normal shape - increased breakdown in the spleen, spleen can also be clogged by them and ischemic ->
- functional asplenism - surface of spleen decreases over time due to clogging by RBC and/or vascular ischemia -> susceptibility to infection increases! especially parvovirus B19
- vascular occlusive crises - acute, episodic painful crises triggered by infections, clod, etc -> MSK pain that is so severe it can require hospital admission. attacks often self-limiting but infarction can occur.
worry about strokes and transient ischemic attacks!
also responsible for “hand-foot syndrome” - dactylitis damaging small bones in hands and feet - swelling
- acute chest syndrome - 30% - dyspnea, chest pain, fever, tachypnea, leukocytosis, pulmonary infiltrate on CXR. caused by vaso-occlusion, infection or pulmonary fat embolus from infarcted marrow.
Investigations for sickle cell anemia?
- sickle cell prep - lower O2 so cells sickle “detects sickling of RBCs under microscope in response to O2 lowering agent”
- Hb electrophoresis to distinguish btwn HbAS (trait), HbSS (homozygous), HbSC (one of S + thalassemias)
- CBC - increased reticulocytes in homozygous and S+thal, decreased Hb
- peripheral blood - sickled cells
- electrophoresis - no HbA, only HbS and HbF, normal HbA2 (no beta there)
Treatment for sickle cell anemia?
- genetic counselling
- folic acid to prevent folic deficiency (want as many RBCs produced as possible, folic acid limiting reactant)
- hydroxyurea to enhance production of HbF (b/c no beta there)
- if vaso-occlusive crisis: give O2, liquid to reduce viscosity, correct acidosis if present (since acidosis or high CO2 can also precipitate), analgesia for pain, transfuse if very serious - acute chest or stroke, multi-organ failure, ICU
- avoid hypoxia, acidosis, dehydration, fever - anothing that leads to sickling, vaccinate in childhood b/c of functinal asplenism
- screen for complications (bloodwork inc CBC, BUN, creatinine), urinalysis for proteinuria and glomerulopathy, transcranial doppler for stroke prevention, retinal exam from 8 yo for retinopathy, echo from 10 yrs for pulmonary hypertension (remember acute chest common)
What are key features of MDS (myelodysplastic syndrome)?
Suspect MDS (myelodysplastic syndrome) if:
- elderly patient
- history of chemo
- gradual onset
- macrocytic anemia
- pancytopenia
- hypolobated neutrophils = 2 lobes vs 3-5N “pseudo-Pelger-Huet neutrophils “
ex. case 78 yo female Hx progressive fatigue, no FHx, clear Abdo, Chest, Cardiac and rectal exams (to rule out blood per rectum), no hematuria, epistaxis (nose bleeds). breast cancer at age 65 (breast exam also negative). no weight loss, fevers, etc - to rule out malignancy.
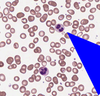
What are Heinz bodies?
Hb gets oxidized and precipitates in G6PD deficiency
Some of it gets removed from RBCs directly by spleen - > bite cells and blister cells
Some of it looks like it precipitated and RBCs have dots -> Heinz bodies (also in alpha thalassemia)
(Oxidative) stress makes me eat bites of fava beans iwth Heinz ketchup
For membrane disorder questions, what is the membrane structure of RBCs?
