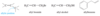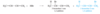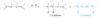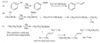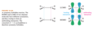Conjugated Systems, Orbital Symmetry, and Ultraviolet Spectroscopy Flashcards
(119 cards)
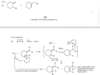
Define:
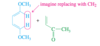
Conjugated
Double bonds can interact with each other if they are separated by just one single bond. Such interacting double bonds are said to be conjugated.
Define:
Isolated Double Bonds
Double bonds with two or more single bonds separating them have little interaction and are called isolated double bonds.

For example, penta-1,3-diene has conjugated double bonds, while penta-1,4-diene has isolated double bonds.
Which is more stable conjugated or isolated double bonds?
Because of the interaction between the double bonds, systems containing conjugated double bonds tend to be more stable than similar systems with isolated double bonds.
What is the heat of hydrogenation of molecules with two isolated double bonds?
When a molecule has two isolated double bonds, the heat of hydrogenation is close to the sum of the heats of hydrogenation for the individual double bonds. For example, the heat of hydrogenation of penta-1,4-diene is -252 kJ/mol (-60.2 kcal/mol), about twice that of pent-1-ene.

What is the head of hydrogenation for conjugated dienes?
For conjugated dienes, the heat of hydrogenation is less than the sum for the individual double bonds. For example, trans-penta-1,3-diene has a monosubstituted double bond like the one in pent-1-ene and a disubstituted double bond like the one in pent-2-ene. The sum of the heats of hydrogenation of pent-1-ene and pent-2-ene is -242 kJ (-57.7 kcal), but the heat of hydrogenation of trans-penta-1,3-diene is only -225 kJ/mol (-53.7 kcal/mol), showing that the conjugated diene has about 17 kJ/mol (4.0 kcal/mol) extra stability.

What happens if two double bonds are even closer together than in the conjugated case?
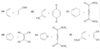
Successive double bonds with no intervening single bonds are called cumulated double bonds. Consider penta-1,2-diene, which contains cumulated double bonds. Such 1,2-diene systems are also called allenes, after the simplest member of the class, propa- 1,2-diene or “allene,” H2C = C = CH2. The heat of hydrogenation of penta-1,2-diene is is -292 kJ/mol (-69.8 kcal/mol), a larger value than any of the other pentadienes.

How stable are cumulated double bonds when compared to the other double bond structures?
Because penta-1,2-diene has a larger heat of hydrogenation than penta-1,4-diene, we conclude that the cumulated double bonds of allenes are less stable than isolated double bonds and much less stable than conjugated double bonds.
Rank each group of compounds in order of increasing heat of hydrogenation.


In a strongly acidic solution, cyclohexa-1,4-diene tautomerizes to cyclohexa-1,3-diene. Propose a mechanism for this rearrangement, and explain why it is energetically favorable.
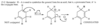
The central carbon atom of an allene is a member of two double bonds, and it has an interesting orbital arrangement that holds the two ends of the molecule at right angles to each other.
(a) Draw an orbital diagram of allene, showing why the two ends are perpendicular.
(b) Draw the two enantiomers of penta-2,3-diene.
What is the name used to describe the extra stability found in conjugated molecules?
This extra stability in the conjugated molecule is called the resonance energy of the system. (Other terms favored by some chemists are conjugation energy, delocalization energy, and stabilization energy.)
Describe the Heat of Hydrogenation of buta-1,3-diene.
The heat of hydrogenation of buta-1,3-diene is about 17 kJ/mol (4.0 kcal/mol) less than twice that of but-1-ene, showing that buta-1,3-diene has a resonance energy of 17 kJ/mol.

What is the most stable configuration of buta-1,3-diene?
The configuartion is planar with the p orbitals on the two pi bonds aligned.
The C2¬C3 bond in buta-1,3-diene (1.48 Å) is shorter than a carbon–carbon single bond in an alkane (1.54 Å). This bond is shortened slightly by the increased s character of the sp2 hybrid orbitals, but the most important cause of this short bond is its pi bonding overlap and partial double-bond character. The planar conformation, with the p orbitals of the two double bonds aligned, allows overlap between the pi bonds. In effect, the electrons in the double bonds are delocalized over the entire molecule, creating some pi overlap and pi bonding in the C2 ¬ C3 bond. The length of this bond is intermediate between the normal length of a single bond and that of a double bond.

Why do we draw plus/minuses on orbitals?
The plus and minus signs used in drawing these orbitals indicate the phase of the wave function, not electrical charges.
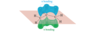
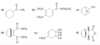
How do the orbitals of ethylene interact with each other?
In the pi bonding molecular orbital of ethylene, the lobes that overlap in the bonding region between the nuclei are in phase; that is, they have the same sign ( + overlaps with +, and - overlaps with -). We call this reinforcement constructive overlap. Constructive overlap is an important feature of all bonding molecular orbitals.
In the pi antibonding molecular orbital (marked by *), on the other hand, lobes of opposite phase (with opposite signs, + with -) overlap in the bonding region. This destructive overlap causes cancelling of the wave function in the bonding region. Midway between the nuclei, this antibonding MO has a node: a region of zero electron density where the positive and negative phases exactly cancel.
Blue = Plus Phase; Green = Minus Phase

What are the energies of the bonding and antibonding MOs?
Electrons have lower energy in the bonding MO than in the original p orbitals, and higher energy in the antibonding MO. In the ground state of ethylene, two electrons are in the bonding MO, but the antibonding MO is vacant. Stable molecules tend to have filled bonding MOs and empty antibonding MOs.
Describe the important principles illustrated in the figure.

Constructive overlap results in a bonding interaction; destructive overlap results in an antibonding interaction. Also, the number of molecular orbitals is always the same as the number of atomic orbitals used to form the MOs. These molecular orbitals have energies that are symmetrically distributed above and below the energy of the starting p orbitals. Half are bonding MOs, and half are antibonding MOs.
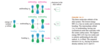
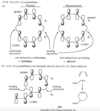
How would you describe buta-1,3-diene’s pi bonding orbitals design (constructive)?
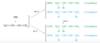
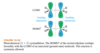
The lowest-energy molecular orbital always consists entirely of bonding interactions. We indicate such an orbital by drawing all the positive phases of the p orbitals overlapping constructively on one face of the molecule, and the negative phases overlapping constructively on the other face. Figure 15-4 shows the lowest-energy MO for buta-1,3-diene. This MO places electron density on all four p orbitals, with slightly more on C2 and C3. (In these figures, larger and smaller p orbitals are used to show which atoms bear more of the electron density in a particular MO.)

How would you describe buta-1,3-diene second pi bonding orbitals design (semiconstructive)?
As with ethylene, the second molecular orbital (π2) of butadiene has one vertical node in the center of the molecule. This MO represents the classic picture of a diene. There bonding interactions at the C1 - C2 and C3 - C4 bonds, and (weaker) antibonding interaction between C2 and C3.

Why would you subtract the number of bonding and antibonding orbitals?
The π2 orbital has two bonding interactions and one anti-bonding interaction, so we expect it to be a bonding orbital (2 bonding - 1 antibonding = 1 bonding). It is not as strongly bonding nor as low in energy as the all-bonding p1 orbital. Adding and subtracting bonding and antibonding interactions is not a reliable method for calculating energies of molecular orbitals, but it is useful for predicting whether a given orbital is bonding or antibonding and for ranking orbitals in order of their energy.

How would you describe buta-1,3-diene’s third pi bonding orbital design (semidestructive)?
The third butadiene MO (π3*) has two nodes (Figure 15-6). There is a bonding interaction at the C2 - C3 bond, and there are two antibonding interactions, one between C1 and C2 and the other between C3 and C4. This an antibonding orbital (*), and it is vacant in the ground state.

How you describe buta-1,3-diene’s fourth pi bonding orbital design (destructive)?
The fourth, and last, molecular orbital (π4*) of buta-1,3-diene has three nodes and is totally antibonding (Figure 15-7). This MO has the highest energy and is unoccupied in the molecule’s ground state. This highest-energy MO (π4*) is typical: For most systems, the highest-energy MO has antibonding interactions between all pairs of adjacent atoms.

Why is buta-1,3-diene is most stable in the planar conformation?
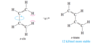
The partial double-bond character between C2 and C3 in buta-1,3-diene explains why the molecule is most stable in a planar conformation. There are actually two planar conformations that allow overlap between C2 and C3. These conformations arise by rotation about the C2 ¬ C3 bond, and they are considered single-bond analogues of trans and cis isomers about a double bond. Thus, they are named s-trans (“single”-
trans) and s-cis (“single”-cis) conformations.
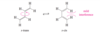
Describe the stabilities between the s-trans and s-cis conformations of buta-1,3,-diene? Against an alkene?
The s-trans conformation is 12 kJ/mol (2.8 kcal/mol) more stable than the s-cis conformation, which whows interference betweent two nearby hydrogen atoms. The barrier for rotation about the C2 - C3 bond from s-trans to s-cis) is only about 29 kJ/mol (about 7 kcal/mol) compared with about 250 kJ/mol (60 kcal/mol) for rotation of a double bond in an alkene. The s-cis and s-trans conformers of butadiene (and all the skew conformations in between) easily interconvert at room temperature.