Cell injury and Necrosis Flashcards
Cell injury
Plasma membrane blebbing, increased intraceullar volume, mitchondrial swelling and calcification, disagregated ribosomes, dilated vesicular ER, aggregated cytoskeletal elements
- several independent cell components are primary tagets for damaging cell stimuli: cell membranes, mitchondria, cytoskeleton, cellular DNA
- damage of one system leads to secondary damage to others
- primary impairment of mitochondrial energy production is due to the lack of oxygen and glucose and toxins like cyanide
- normal concentration of calcium in cytosol is very low, free calcium is used by secondary messenger systems to activate various cytosolic enzymes(protein kinases, phospholipases, calpain)
- calcium is rapidly removed from cytosol by ATP dependent calcium pumps and next bound to buffering proteins
- if Ca2+ is not buffered or pumped out of cells, uncontrolled enzyme activation
- in all cells, reactive oxygen metabolites are constantly generated and can be potentially damging to cells, so they ae constantly scavenged by antioxidants, glutathione peroxidase, supeoxide dismutase and catalse
- ROS is generated when oxygen is reduced to water and most are created by systems involved in electron and oxygen transport
Common causes of cellular injury
(factors of cellular injury)
-
Hypoxia and ischemia:
- Hypoxia, which refers to oxygen deficiency, and ischemia, which means reduced blood supply, are among the most common causes of cell injury. Both deprive tissues of oxygen, and ischemia and results in a deficiency of essential nutrients and a build up of toxic metabolites. The most common cause of hypoxia is ischemia resulting from an arterial obstruction, but oxygen deficiency also can result from inadequate oxygenation of the blood, as in a variety of diseases affecting the lung, or from reduction in the oxygen-carrying capacity of the blood, as with anemia of any cause, and carbon monoxide (CO) poisoning.
- Toxins. are encountered daily in the environment; these include air pollutants, insecticides, CO, asbestos, cigarette smoke, ethanol, and drugs. Many drugs in therapeutic doses can cause cell or tissue injury in a susceptible patient or in many individuals if used excessively or inappropriately. Even innocuous substances, such as glucose, salt, water and oxygen, can be toxic. Infectious agents. All types of disease-causing pathogens, including viruses, bacteria, fungi, and protozoans, injure cells.
- Immunologic reactions: Although the immune system defends the body against pathogenic microbes, immune reactions also can result in cell and tissue injury. Examples are autoimmune reactions against one’s own tissues, allergic reactions against environmental substances, and excessive or chronic immune responses to microbes . In all of these situations, immune responses elicit inflammatory reactions, which are often the cause of damage to cells and tissues.
- Genetic abnormalities. Genetic aberrations can result in pathologic changes as conspicuous as the congenital malformations associated with Down syndrome or as subtle as the single amino acid substitution in hemoglobin giving rise to sickle cell anemia. Genetic defects may cause cell injury as a consequence of deficiency of functional proteins, such as enzymes in inborn errors of metabolism, or accumulation of damaged DNA or misfolded proteins, both of which trigger cell death when they are beyond repair.
- Nutritional imbalances. Protein–calorie insufficiency among impoverished populations remains a major cause of cell injury, and specific vitamin deficiencies are not uncommon even in developed countries with high standards of living. Ironically, excessive dietary intake may result in obesity and also is an important underlying factor in many diseases, such as type 2 diabetes mellitus and atherosclerosis.
- Physical agents. Trauma, extremes of temperature, radiation, electric shock, and sudden changes in atmospheric pressure all have wide-ranging effects on cells
What is cell death?
- inability to adapt to an environmental change leads to failure of cellular function and may result in sublethal cellular damage or death
- 2 kinds of death: necrosis and apoptosis

What is apoptosis?
- programmed cell death
- endogenous programed and energy dependent process designed to specifically switch off cells or eliminate them
- occurs when a cell dies through the activation of internally controlled suicide program

What are physiological, adaptive and pathological events in apoptosis?
- programmed destruction of cells during embryogenesis
- hormone dependent involution in the adult(endometrial cell breakdown, regression of lactating breast after weaning, prostatic atrophy after castration)
- cell deletion in proliferating cell population
- cell death in tumor
- death of neurophils
- cell death induced by cytotoxic t cells
- pathologic atrophy in parenchymal organs after duct obstruction
- cell injury in certain viral diseases(adenovirus, HIV infections)
- cell death produced by injurious stimuli, which are given in low doses, large doses of same stimuli result in necrotic cell death(heat, radiation, cytotoxic anticancer drugs, hypoxia)
What are the energy dependent cascade of molecular events for apoptosis?
- signaling pathways: transmembrane may be negative or positve determinants of apoptosis
- apoptosis is activated by caspases which exist as inactive proenzyme and must undergo enzymatic cleavage to become active
- two distinct pathways lead to caspases activation:
- mitochondrial / intrinsic pathway: Bcl-2 family of proteins- major mechanism in mammilian cells
- death receptor/extrinsic pathway: Fas-Fas ligand model, TNF receptor
- apoptotic signals result in mitochondrial permeability transitions. Formation of pores within the inner miochondrial membrane results in reduction of membrane potential, cytochrome c is releasing from mitochondria to cytosol(ativates caspases apoptosis initiating factor-AIF)
- Bcl-2 and Bcl-XL can suppress apoptosis(prevent cytochrome c release and inhibit Apaf-1 induced caspase activation
- BAX and BAK are pro-apoptotic
Apoptosis- Microsopic view
What are the phases?
- First phase priming
- normal cells are arranged in close contact and are united by cell junctions
- synthesis of enzymes can cause cell dissolution, not associated with structural changes
- in development many cells are primed for apoptosis and survive if rescued by specific trophic factor
- Second phase- cell excutioner pathway
- apoptotic cells lose surface specializations and junctions, shrinking in size, nuclear chromatin condenses but cell organelles remain normal
- endonuclease enzymes cleave chromosomes into individual nucleosome fragments
- Third phase- degradation
- splinting of cell into several fragments called apoptotic bodies
- nuclear fragmentation occurs
- each fragment contains viable mitochondria and intact organelles
- takes a few minutes
- caspases, specifically proteases are the main enzymes
- fourth phase: phagocytosis
- apoptotic fragments are recognized by adjacent cells, which inges them by phagocytosis for destuction
- some fragments degenrate extracellularly, while others are ingested by local phagocytic cells
What is oxidative stress?
- Oxidative stress refers to cellular abnormalities that are induced by ROS, which belong to a group of molecules known as free radicals.
- Free radical-mediated cell injury is seen in many circumstances, including chemical and radiation injury, hypoxia, cellular aging, tissue injury caused by inflammatory cells, and ischemia-reperfusion injury. In all these cases, cell death may be by necrosis, apoptosis, or the mixed pattern of necroptosis.
- Free radicals are chemical species with a single unpaired electron in an outer orbit. Such chemical states are extremely unstable, and free radicals readily react with inorganic and organic molecules; when generated in cells, they avidly attack nucleic acids as well as a variety of cellular proteins and lipids.
- In addition, free radicals initiate reactions in which molecules that react with the free radicals are themselves converted into other types of free radicals, thereby propagating the chain of damage.
The generation of free radicals is increased under several circumstances:
- The absorption of radiant energy (e.g., ultraviolet (UV) light, x-rays). Ionizing radiation can hydrolyze water into hydroxyl (•OH) and hydrogen (H•) free radicals.
- The enzymatic metabolism of exogenous chemicals (e.g., carbon tetrachloride)
- Inflammation, in which free radicals are produced by leukocytes
- Reperfusion of ischemic tissues, as already described.
Explain Generation and Removal of Reactive Oxygen Species
- The accumulation of ROS is determined by their rates of production and removal ROS are produced by two major pathways.
- ROS are produced normally in small amounts in all cells during the reduction-oxidation (redox) reactions that occur during mitochondrial respiration and energy generation. In this process, molecular oxygen is reduced in mitochondria to generate water by the sequential addition of four electrons. This reaction is imperfect, however, and small amounts of highly reactive but short-lived toxic intermediates are generated when oxygen is only partially reduced. These intermediates include superoxide (O2 • ), which is converted to hydrogen peroxide (H2O2) spontaneously and by the action of the enzyme superoxide dismutase (SOD). H2O2 is more stable than O2 • and can cross biologic membranes. In the presence of metals, such as Fe2+ , H2O2 is converted to the highly reactive hydroxyl radical •OH by the Fenton reaction.
- ROS are produced in phagocytic leukocytes, mainly neutrophils and macrophages, as a weapon for destroying ingested microbes and other substances during inflammation and host defense. The ROS are generated in the phagosomes and phagolysosomes of leukocytes by a process that is similar to mitochondrial respiration and is called the respiratory burst (or oxidative burst). In this process, a phagosome membrane enzyme catalyzes the generation of superoxide, which is converted to H2O2. H2O2 is in turn converted to a highly reactive compound, hypochlorite (the major component of household bleach), by the enzyme myeloperoxidase, which is present in leukocytes.
- Nitric oxide (NO) is another reactive free radical produced in macrophages and other leukocytes. It can react with O2 − to form a highly reactive compound, peroxynitrite, which also participates in cell injury
Cells have developed mechanisms to remove free radicals and thereby minimize their injurious effects. How do cells remove free radicals?
Cells have developed mechanisms to remove free radicals and thereby minimize their injurious effects. Free radicals are inherently unstable and decay spontaneously. There also are nonenzymatic and enzymatic systems, sometimes called free radical scavengers, serving to inactivate free radicals
- The rate of decay of superoxide is significantly increased by the action of superoxide dismutase (SOD).
- Glutathione (GSH) peroxidases are a family of enzymes whose major function is to protect cells from oxidative damage. The most abundant member of this family, GSH peroxidase 1, is found in the cytoplasm of all cells. It catalyzes the breakdown of H2O2 by the reaction 2GSH + H2O2 → GS-SG + 2H2O. The intracellular ratio of oxidized GSH to reduced GSH is a reflection of this enzyme’s activity and thus of the cell’s ability to catabolize free radicals.
- Catalase, present in peroxisomes, catalyzes the decomposition of hydrogen peroxide (2H2O2 → O2 + 2H2O). It is one of the most active enzymes known, capable of degrading millions of molecules of H2O2 per second.

Explain generation of reactive oxygen metabolites in cell injury
- free ion
- xanthine accumulates in hypoxic tissues as a metabolite of ATP. In hypoxic conditions accumulated xanthine can be oxidized by xanthine oxidase
- following ischemia, cells become depleted of energy, but ROS does not develop because no oxygen in tissues, If tissues are reperfused, huge amounts of reactive oxygen are generated
- tissue necrosis occurs not due to cessation of blood supply but reestablishment
ROS causes cell injury by damaging which components?
ROS causes cell injury by damaging multiple components of cells:
- Lipid peroxidation of membranes. Double bonds in membrane polyunsaturated lipids are vulnerable to attack by oxygen-derived free radicals. The lipid–radical interactions yield peroxides, which are themselves unstable and reactive, and an autocatalytic chain reaction ensues. Damage to plasma membranes as well as mitochondrial and lysosomal membranes can have devastating consequences, as discussed earlier in the context of ischemia and hypoxia.
- Crosslinking and other changes in proteins. Free radicals promote sulfhydryl-mediated protein crosslinking, resulting in enhanced degradation or loss of enzymatic activity. Free radical reactions also may directly cause polypeptide fragmentation. Damaged proteins may fail to fold properly, triggering the unfolded protein response, described later.
- DNA damage. Free radical reactions with thymine residues in nuclear and mitochondrial DNA produce singlestrand breaks. Such DNA damage has been implicated in apoptotic cell death, aging, and malignant transformation of cells.
- In addition to the role of ROS in cell injury and the killing of microbes, low concentrations of ROS are involved in numerous signaling pathways in cells and thus in many physiologic reactions. Therefore, these molecules are produced normally but, to avoid their harmful effects, their intracellular concentrations are tightly regulated in healthy cells.
Calcium stores issues low chart

Free radicals flow chart

What is necrosis?
- necrosis is a common type of cell death after exogenous stimuli occuring after such stresses like ischemia after chemical injury
- oncosis is prelethal changes preceding necrotic cell death, characterized by cellular swelling and can be distinguisehd from prelethal changes in apoptosis, associated largely wit cellular shrinkage
- if damage to the cell is minimal, the cell can recover following removal of the damaged stimulus. In other situtaions, a damaging stimulus may be sublethal and cell cannot recover leading to cell death
- damaging stimulus to a cell is massive, the cell is killed immediately without passing through stages of necrosis
- Necrosis is a form of cell death in which cellular membranes fall apart, and cellular enzymes leak out and ultimately digest the cell and occurs due to underlying pathlogical process
- Necrosis elicits acute inflammation(bacterial or necrotic- MI-neutrophils)that is induced by substances released from dead cells and which serves to eliminate the debris and start the subsequent repair process.
- The enzymes responsible for digestion of the cell are derived from lysosomes or from the dying cells or from leukocytes recruited as part of the inflammatory reaction.
- Necrosis often is the culmination of reversible cell injury that cannot be corrected.
- The mechanisms of necrosis include: failure of energy generation in the form of ATP because of reduced oxygen supply or mitochondrial damage; damage to cellular membranes, including the plasma membrane and lysosomal membranes, which results in leakage of cellular contents including enzymes; irreversible damage to cellular lipids, proteins, and nucleic acids, which may be caused by reactive oxygen species (ROS).
What are the cellular events in necrosis?
- many changes are caused by lysosomal hydrolases which are released into the cell when cell membrane integrity is lost
- intense eosinophilia of the dead cell is due to the loss of RNA and coagulation of proteins
- nuclei undergoes pyknosis, karyorrhexis, karyolysis leaving a shrunken cell devoid of a nucleus
- proteins may be liberated from the dead cells and be detected in blood in diagnosis
- Leakage of intracellular proteins through the damaged cell membrane and ultimately into the circulation provides a means of detecting tissue-specific necrosis using blood or serum samples. Cardiac muscle, for example, contains a unique isoform of the enzyme creatine kinase and of the contractile protein troponin, whereas hepatic bile duct epithelium contains the enzyme alkaline phosphatase, and hepatocytes contain transaminases. Irreversible injury and cell death in these tissues elevate the serum levels of these proteins, which makes them clinically useful markers of tissue damage.

What is coagulative necrosis?
- necrotic tissue that appears firm and pale as if cooked
- cellular outline and tissue architecture can be discerned histologically, nucleus disappears and increased eosinophilic cells that lasts for days or weeks. Cell shape and organ structure are preserved by coagulation of cellular proteins.
- Leukocytes are recruited to the site of necrosis, and the dead cells are ultimately digested by the action of lysosomal enzymes of the leukocytes. Cells have relatively few lysosmes to bring about complete breakdown of cellular proteins
- The cellular debris is then removed by phagocytosis mediated primarily by infiltrating neutrophils and macrophages.
- most common cause: occlusion of arterial supply to a tissue. Characteristic of ischemic infarction anywhere except the brain which dentaures enzymes and blocks proteolysis
- proteins liberated from the dead cell can enter the blood
- ex: MI
- Ex: area of infarcted tissue is often wedge shaped and pale(kidney). Main vessel with branching vessels and thrombus in the middle. Above occlusion is infarct
- Ex: Red infarction if blood reenters loosely organised tissue(red testicle- collapsed vein and blood reenters through a. but v. cannot take out the blood so blood builds up)
Describe acute myocardial infarction
(Coagulative necrosis)
- regional MI (90% of cases) involves one segment of ventricular wall
- Cause: thrombus formation on a complicated atheromatous plaque, if occulsion is complete then the infarct is full in thickness, if there is lysis of the thrombus or a collateral supply to the myocardium, the infarct will be limited to the subendocardial zone
- circumferential subendocardial infarction(10%) of cases involves the subendocardial zone of ventricle
- cause: hypoperfusion of the main coronary arteries (high grade atheresclerotic stenosis)
What are the 3 patterns of myocardial infarction?
- Regional Full thickness (complete persistent thrombotic occlusion, ex: full thickness of lateral wall of left v. is infarcted)
- regional subendocardial(flow reestablished after occlusion)
- circumferential subendocardial (severe reduction of lumen in main arteries, ex: the subendocardial zone around the whole circumference of left v. is infarcted and dark in color)
Describe appearances of myocardial infarction
- between 0-12 hours, an infarct is not microscopically visible, the ischemic muscle can be detected by showig a loss of oxidative enzymes(NBT)- the infarcted area appears uncolored
- between 12 and 24 hours, he infarcted area is microscopically pale, histologically infarcted muscle is brightly eosinophilic with intercellular edema
- between 24 and 72 hours, the infarcted area excites in actue inflammatory response(macro-soft and pale with a slight yellow color), histologically neutrophils infiltrate between dead cardiac muscle fibers
- between 3-10 days- organization in infarcted area (macro-hyperaemic border develops around the yellow dead muscle)- histologically - vascular granulation tissue
- after weeks or months the infarct is replaced by collagenous scar
Myocardial infarct - arteries
the coronary a. shown has narrowing of the lumen due to build up of atherosclerotic plaque. Severe narrowing can lead to angina, ischemia and infarction

Myocardial infarct- arteries
Distal portion of coronary a. shows significant narrowing. Such distal involement is typical of severe coronary atherosclerosis such as can appear with diabetes mellitus or famiial hypercholestorelemia. This makes coronary bypass operation difficult

Myocardial infarct
The left ventricular wall can have large MI. The center of the infarct contains necrotic muscle that appears yellow-tan. Surrounding this zone of red hyperemia. Remaining viable mycocardium is reddish brown
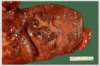
Myocardial infarct
Left ventricle of heart, extending from anterior portion and into septum is a large recent mycoardial infarction. The center is tan with surrounding hyperemia. The infarction is transmural because it extends through the full thickness of the wall

Myocardial infarct: histological
Earliest change seen in first day is contraction band necrosis. The mycoardial fibrils are beginnig to lose cross striations and the nuclei are not clear. Many irregular darker pink wavy contraction extending across the fibers

Mycardial infarct: histological
the myocardial fibers have dark red contraction bands extending across them. The myocardial cell nuclei have almost all disappeared. There is beginning of acute inflammation

Early myocardial infarction

- Acute inflammatory cell infiltrate and the myocardial fibers are so necrotic that the outlines of them are only barely visible.
- Dark red contraction bands.
- The myocardial cell nuclei have all disappeared.



























































