A1-2: Pharmacodynamics Flashcards
Difference between pharmacodynamics and pharmacokinetics?
Pharmacodynamics: how drug acts on the body (therapeutic effects, side effects)
Pharmacokinetics: how body responds to the drug (absorption, distribution, metabolism, excretion)
What are the broad categories that can be considered targets of drugs?
- Membrane receptors (GPCRs, TKs, Ligand-gated ion channels)
- Intracellular receptors (steroid receptors, nuclear receptors, PPAR-gamma)
- Voltage-gated ion channels (e.g. calcium channel blockers)
- Enzymes: usually inhibited (e.g. statins, aspirin, MAOI)
- Transporters (e.g. SSRI/SNRIs, cocaine)
- Structural proteins (e.g. integrin antagonist to disrupt T cells in a MS drug)
- Nucleic Acids (e.g. cancer drugs)
- Assorted atypical drugs: antacids (provide base), activated charcoal (binds to prevent absorption)
(“VIETNAMS” may work as a mnemonic)
What are some examples of receptors that are coupled to G proteins (GPCRs)?
- Adrenergic: α1: Gq, α2: Gi, ß1: Gs, ß2: Gs, ß3: Gs (QISSS)
- Muscarinic: M1: Gq, M2: Gi, M3: Gq (QIQ)
- Dopamine: D1: Gs, D2: Gi
- Histamine: H1: Gq, H2: Gs
- Vasopressin: V1: Gq, V2: Gs
- GABA-B: Gi (GABA-A is ionotropic)
- ACTH
- PTH
- Glycoprotein receptors (FSH, LH, TSH)
- Most neuropeptide receptors (Opioid, Angiotensin, Bradykinin, CCK, NPY, Oxytocin, etc)
(there are more but this is already enough for one card, just trying to remember some important examples)
What are the 3 classes of GPCRs?
- Class I: Rhodopsin-Like
- Class II: Secretin Receptor-Like
- Class III: Metabotropic Glu-Like
What are some enzymes that are commonly the target of drugs?
- COX (e.g. Aspirin)
- ACE (e.g. enalapril)
- Acetylcholinesterase (e.g. neostigmine)
- MAO (e.g. moclobemid, selegiline)
- COMT (e.g. entacapon)
- HMG-CoA reductase (e.g. atorvastatin)
- Dihydropholicacid-reductase (e.g. methotrexate)
- GABA-transaminase (e.g. vigabatrin)
What are some examples of transport proteins that are the target of drugs?
- Serotonin transporter (e.g. citalopram)
- Noradrenalin transporter (e.g. reboxetin)
- Dopamine transporter (e.g. cocaine, bupropion)
- Vesicular monoamine transporter (tetrabenazine)
What are some voltage-dependent ion channels that are the target of drugs?
-
Sodium channels:
- Local anesthetics e.g. lidocaine
- Antiepileptics e.g. carbamazepine
- Antiarrhythmics e.g. chinidine
-
Potassium channels
- Antiarrhythmics e.g. sotalol, amiodarone
- Any drug that prolongs the QT time (some antipsychotics, macrolide antibiotics, methadone, antihistamines)
-
Calcium channel:
- L-type e.g. dihidropyridines like amlodipine
- T-type e.g. ethosuximide
- N-type e.g. gabapentin
What is receptor theory?
A drug works by acting as a ligand on a specific receptor.
Receptors are typically specific, saturatable, and finite in number
A drug is different than for example a heavy metal, which just binds to whatever -SH groups it can and denatures proteins.
How do you calculate the dissociation constant of a drug that reversibly binds to a receptor?
What unit is it expressed in?

How does the dose-response curve (max binding vs ligand concentration) change if the ligand concentration is expressed in a logarithmic scale?
Max binding vs [Ligand]: hyperbolic curve
Max binding vs log[Ligand]: sigmoid curve

What is the difference between potency and efficacy, and how is this difference expressed on a graph?
Potency: amount of drug necessary to produce an effect of a given magnitude. Based on the EC50 (quantity producing 50% of maximum effect)
Efficacy: magnitude of response a drug causes when it interacts with a receptor. Dependent on # of drug-receptor complexes + intrinsic activity of drug. Based on Emax.

What is an agonist, and what are the different types of agonists?
Agonist: binds a receptor and then produces a biological response based on the agonist concentration and amount of activated receptors
- Full Agonist: evokes maximal effect possible (e.g. NE on α1R)
- Partial Agonist: evokes less-than-maximal effect even with 100% receptor occupancy (e.g. pindolol on ßR)
- Inverse Agonist: bind same receptor as agonist but decrease constitutive activity of the receptor, decreasing or antagonizing the effects of an agonist (e.g. metoprolol on ßR)

What is an antagonist?
What are the different types of antagonists?
Antagonists bind a receptor but does not change effects from the constitutive receptor activity (hence aka “neutral antagonist”) - has no effect in the absence of an agonist
- Competitive Antagonist: agonist and antagonist bind to same site reversibly (e.g. terazosin competes with NE in α1R) - [note that partial agonists may function as competitive antagonists when given with full agonists]
- Irreversible Antagonist: Covalently bind active site of receptor (often not used in pharma but good example is aspirin, which eventually loses anticoagulant effect bc of platelet turnover)
- Allosteric Antagonist: binds different site than agonists but still modifies target, causing reduction of Emax without change in EC50 (e.g. picrotoxin binding GABA receptor -> impossible for Cl- to pass through)
- Functional Antagonism: i.e. receptor X and receptor Y exert opposite reactions, and drug A acts on receptor X, while drug B acts on receptor Y, similar to sympathetic vs parasympathetic activation (e.g. Epinephrine vs Histamine in bronchodilation vs bronchoconstriction)
Both allosteric and function antagonists show “non-competitive” nature in the graph

What does the “quantal dose effect” or “cumulative dose-response curve” graph look like?
(it’s the graph showing both ED50 and LD50, and part of the “relation between drug dose and clinical response” part of the topic list)

What is therapeutic index, and how do you calculate it?
What is therapeutic window?
TI is the ratio of the dose that produces toxicity in half the population (TD50) to the dose that produces the clinically-desired / effective response in half the population (ED50). More for animal studies, or use modified for human studies (Riba gave us some different ways to put it but this is from the book)
TI = TD50 / ED50 (higher TI = safer)
Therapeutic Window is the range between minimum toxic dose and the minimum therapeutic dose (reflects dosages that are both safe and effective; has greater practical value)
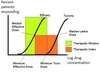
What are 4 examples of drugs with a low TI?
Warfarin
Theophylline
Digoxin
Lithium
(WTDL: Warning: These Drugs are Lethal)
- What is it called when a drug exerts a diminished response over the course of therapy?
- What is it called if that response diminishes rapidly after drug administration?
- Tolerance
- Tachyphylaxis
(Extra: what is colloquially “low tolerance” would be called “hyper-reactive.” The term “hypersensitivity” should be used for allergic responses)
What are 2 mechanisms of increasing drug tolerance?
- Decreasing number of receptors (down-regulation)
- Receptor-ligand coupling becoming inefficient (desensitization)
What are the possible pharmacodynamic drug interactions + some examples?
- Synergistic Good: enhance each others’ effects in beneficial way (e.g. ACE Inhibitors + Thiazide diuretics reduce hypertension)
- Synergistic Bad: enhance each others’ effects too strongly or in harmful way (e.g. ACE inhibitors + thiazidie now causing hypotension; or Warfarin + aspirin increasing risk of bleeding)
- Antagonistic Good: opposite effects that help the patient (e.g. ACE inhibitors cause hyperkalemia, Thiazide causes hypokalemia. Together they are more balanced)
- Antagonistic Bad: opposite effects that are harmful (e.g. bactericidal + bacteriostatic antibiotics. Bacteriostatic ATBs inhibit effects of bactericidal ATBs)
