5E: principles of chemical thermodynamics & kinetics (all GC) Flashcards
Thermodynamics laws: game analogy
- Zeroth Law*
- First Law*
- Second Law*
- Third Law*
- Zeroth law: says temperature exists and it can equilibriate, sets ground rules for the game
- First law: says change in energy always = sum of heat and work, says you can’t win, conservation of energy in thermodynamic processes
- Second law: says temperature and pressure flow downhill from greater to less and that net entropy of the universe is always increasing, says you can’t even break even
- Third law: says absolute zero is untenable since zero energy can’t be achieved, says you can’t even end the game
Thermodynamic system-state function & path function
- State function: properties that describe the current state of the system. Not affected by how the systems got to their state, just the properties of their current state (ex: temperature, pressure, volume)
- Path function: properties that depend on the pathway used to achieve a state (ex: work, heat)
- Thermodynamic systems can’t describe systems on a molecular scale. Thermodynamic systems average out all molecular interactions to find an average. Molecular scale means the sample size is too small
- Internal energy: collective energy of molecules measured on a microscopic scale. Many different types!
Internal Energy Types
- Vibrational energy: created by back-and-forth motion of atoms
- Rotational energy: created by rotation of a molecule around its center of mass
- Translational energy: created by movement of a molecule’s center of mass
- Electronic energy: potential electric energy created by attractions between electrons and their nuclei
- Intermolecular potential energy: intermolecular dipole forces
- Rest mass energy: described by E = mc^2
What are the 2 ways to transfer energy between systems?
- heat & work
- Difference between heat and work: directional collisions vs. random collisions
-
Work is done by energy transfer through _ordered molecular collisions_
- more constrainsed the molecules are (higher the P, lower the V)=greater capacity to do work
- Heat is done through _random collisions_ between high energy & low energy particles
Zeroth Law
- Concept of temperature
- temperature: thermal energy per mol of molecules
First Law of Thermodynamics
-
conservation of energy in thermodynamics
- total energy of system & surroudings always conserved
-
ΔE = q + w
- Energy change of a system = heat flow into the system + work done on the system
-
For closed systems, only internal energy change takes place, ΔU is used instead of ΔE
- If no change in volume occurs as well, omit work. ΔU = q
- Energy transferred out of a system during expansion (ΔE negative)
- Energy transferred into a system during contraction (ΔE positive)
PV Diagram:
Work
- Work done = area under or enclosed by curve
- Work= any energy transfer that’s not clear
- System performs PV work by changing it’s size or shape using energy from the system
- Ex: piston expanding
- Formula for PV work: W= PΔV
- Volume must be constant
- Note how negative work=work done by the system (if V expands)
-
Positive work=work done ON THE SYSTEM
- Keep in mind the MCAT might try to define work done by the system as positive
-
PV diagram: x-axis=P, y-axis=V
- Work=area under the PV curve
- Work is a path function, different curve/path results in a different
amount of work
Second Law: concept of entropy
- ΔSsystem + ΔSsurroundings = ΔSuniverse
-
Entropy as a measure of “disorder”
- Entropy: energy trying to spread itself evenly throughout the universe
- Entropy of an isolated system will never decrease. Since the universe is an isolated system, entropy of the universe never decreases
- Rxn must increase entropy of the universe to proceed
- Entropy increases with number, size, volume, and temperature
- Relative entropy for gas, liquid, and crystal states
Third Law: absolute zero
- Says zero entropy can only take place at absolute zero.
- However, this is unattainable so third law can only be realized in theory
- Measurement of heat changes (calorimetry)
- What are the two types of calorimetry?
- Calorimetry: measuring changes in heat flow of rxn by monitoring temp change of a calorimeter coupled to the rxn to find the change in enthalpy
-
Two types: constant pressure and constant volume
-
Coffee cup calorimeters (constant pressure)
- Rxn takes place in chamber with open top. Constant pressure of local atmosphere dictates pressure
- Use insulated chamber to prevent heat exchange w/ surroundings
- Used to measure heats of reaction and enthalpy b/c no heat from rxn is lost to surroundings
-
Bomb calorimeter (constant volume):
- Rxn takes place in rigid, sealed off container
- Use insulated chamber to prevent heat exchange with
-
Coffee cup calorimeters (constant pressure)
surroundings
* Measures internal energy change by finding q **from q =**
CΔT
* *C of calorimeter is known, T change can be* * measured after the reaction* * *important for calorimeter chamber to be thermally insulated from the surroundings. No heat exchange between system & surroundings* *
- Heat Capacity, what gives greater heat capactiy?
- Specific Heat Capacity
- Heat capacity: How much E must be added to a substance to change its temp by 1 C or K
- More bonds in a molecule = greater heat capacity
- This is b/c E is redirected to stretching these bonds instead of raising T
- More IMF’s between molecules = greater heat capacity
- This is b/c IMF’s must be broken using E to raise temperature. Some E has to be redirected to do this
- T will always increase when E is added to a substance at a constant V and P
- Formula for heat capacity: q = mcΔT
-
Specific heat capacity: intrinsic property, heat capacity per unit mass
- Q = mcΔT
- ΔHrxn = -ΔHcalorimeter
Heat transfer:
1. Conduction:
2. Convection
3. Radiation
-
Conduction: heat transfer through molecular collisions. Requires direct physical contact
- Thermal conductivity (k): an object’s ability to conduct heat, depends on composition and temperature (composition more so)
-
Convection: heat transfer through fluid
- Driven by differences in pressure and density, drives warm fluid in direction of cooler fluid
- Ex. Hot air rises, causing cooler air from ocean to move in
-
Radiation: thermal energy transfer via electromagnetic waves
- Newton’s law of cooling: a body’s rate of cooling is proportional to the temperature difference between a body and it’s environment
-
Emissivity: fraction of radiation absorbed by a surface (depends on surface composition)
- Higher emissivity = higher amount of radiation absorbed
Endothermic/Exothermic reactions
Enthalpy (ΔH)
-
Enthalpy: Used to measure changes in heat. Sum of internal energy and work.
- ΔH=ΔU+PΔV
- H = enthalpy
- U = internal energy
- P = pressure, V = volume
-
When only PV work is done at constant pressure and volume, ΔH = q
- This is b/c no non=PV work means PΔV value is 0
- Standard enthalpy of formation (Hfo):
- standard state
- postive & negative Hfo change
- Standard enthalpy of formation (Hfo): change in heat for a reaction that creates one mole of that compound from its raw elements in their standard states
- Standard state: reference form of a substance
- Positive Hfo change = endothermic reaction, heat flows into
system
- Negative Hfo change = exothermic reaction, heat flows out of
system
Hess’ Law of Heat Summation
- Sum of enthalpy changes for each step equals total energy change
- Forward reaction has the opposite change in enthalpy as reverse
-
Energy reaction diagram
- Y-axis:“energy” can be enthalpy, Gibbs, or energy
- x-axis: rxn progresses
- Difference between initial and final energy states is constant regardless of changes in activation energy
Gibbs Free energy: ΔG
- way of finding entropy change in both system and surroundings using only information about the system,
-
ΔG = ΔH – TΔS
- ΔG shows how much non-PW work is “free” for a reaction, determines if a reaction is spontaneous or not
- ΔG, ΔH, and ΔS only refer to changes in the SYSTEM, not the surroundings
- Both ΔS and ΔH are required in determining if a reaction proceeds spontaneously
- G is an extensive property, state function (like enthalpy, entropy)
Spontaneous reactions and ΔGo
- Negative ΔGo means a reaction proceeds spontaneously
- ΔGo value depends on both ΔH and ΔS
Heat of fusion, heat of vaporization
-
Heat of vaporization is usually greater than heat of fusion
- Usually break more bonds from liquid to gas than solid to liquid
Phase Diagram: pressure & temperature
Critical Temperature
Critical pressure
Critical Point
- Phase diagram indicates phases of a substance at different pressures and temperatures
-
Critical temperature: temperature at which a substance cannot be liquefied regardless of pressure
- Critical pressure: pressure at the critical temperature
- Critical point: point defined by the intersection of the critical T and P
- Solids and gases favored at extreme conditions, liquids favored in intermediate conditions
-
Difference in water’s phase diagram
- Line between solid and liquid phase has NEGATIVE slope. This allows solid water to be less dense than liquid water
- Normally, this line has a POSITIVE slope
Rate law, rate constant
- Rate law: finds the rate of a reaction by factoring in rate law and concentrations of reactants
- Experimentally determining rate law:
- Compare reaction rates in two trials where concentrations of all reactants except for one stay constant
- Compare how changing concentration of one reactant affects reaction rate
* If reactant A is doubled and rate doubles, then order of reactant A is one. If A is doubled and rate quadruples, then order is two
- Compare how changing concentration of one reactant affects reaction rate
Reaction Order
- Zero Order*
- First Order*
- Second/Third Order*
- Rateforward = k[A]a[B]b, rate law = a + b
-
Zero order: rxn rate is independent of concentration of any reactant
- Graph: [A] over t, slope of –k is linear and downwards
-
First order: rxn rate is directly proportional to concentration of a single reactant
- Graph: ln[A] over t, slope of –k is linear and downwards
- Second/Third order: rxn rate is proportional to a single reactant’s concentration raised to the second/third power OR the product of
- concentrations of multiple reactants
Rate-determining Step
- In multistep reactions, rate of the slowest occurring step is the rate determining step
- Rate-determining step determines the rate law for the reaction
Dependence of reaction rate upon temperature
Activation Energy
-
Requirements to reach activation E
- 1) Particles must be traveling at a sufficient velocity
- 2) Particles must collide in the correct orientation
- Activated complex or transition state
- Interpretation of energy profiles showing energies of reactants, products, activation energy, and ΔH for the reaction
Dependence of reaction rate upon temperature
Use of Arrhenius Equation
-
Used to find rate constant (k). Rate constant dictates rate of reaction (directly proportional)
- If it affects rate constant, it affects rate of reaction in the same way
- Rate constant is inversely proportional to activation energy, directly proportional to temperature and collision frequency
- p = steric factor, takes into account correct orientation of molecules hitting each other and frequency of collisions
What are the 3 factors that affects K in the Arrehius Equation?
- 1) Pressure: higher pressure increases rate constant (relevant for gases)
- 2) Catalysts: presence of catalyst increases rate constant
- 3) Temperature: temperature increases rate constant
Kinetic control versus thermodynamic control of a reaction
-
Kinetic control: controls speed of a reaction, governed by Ea
- Pertains to top half of a reaction energy diagram (where transition state peak is)
-
Thermodynamic control: controls whether or not a reaction will occur, governed by ΔG of the reaction
- Pertains to bottom half of reaction energy diagram
Catalysts
What are the two types of catalysts?
Total rate law of catalyzed reactions:
- If concentration of catalyst far outweighs reactant, then rxn order is zero
- Increases rates of forward and reverse reactions
- Can lower Ea, increases steric factor, or both
- However, does not affect overall change in energy, difference between final energies of reactants and products stays the same, only the Ea changes
Two types: heterogeneous and homogenous
-
Heterogeneous: different phase than reactants of products. Rate of catalysis depends on strength of attraction between reactant and catalysts (ex: liquid catalyst adsorbs, or sticks to, a solid reactant). Rxn rate enhanced by increasing surface area of catalyst
- Too little attraction: catalyst can’t adsorb to reactant, little effect on reaction rate
- Too much attraction: catalyst doesn’t want to let go, little effect on reaction rate
- Want to get just the right amount
- Homogeneous: same phase as reactants and products
-
Total rate law of catalyzed reactions = rate law of original reaction + rate law of catalyzed reaction
- Ex: Rate law of acid catalyzed reaction = k0[A] + kH+[H+][A]
- B/c even with catalyst, original reaction will still occur
Equilibrium in reversible chemical reactions
Law of Mass Action
Equilibrium Constant Keq
Reaction Quotient (Q)
proportionals Keq & Q
-
Law of Mass Action
- Rate of any reaction is proportional to the concentrations of the
different species in the reaction (according to the equilibrium constant)
- Equilibrium Constant (see pic attacked for equation)
-
Reaction Quotient (Q)
- Same formula as equilibrium constant but reaction isn’t necessarily at equilibrium
-
Value of Q relative to Keq can tell us about _direction rxn will proceed:_
- Q = K: rxn is at equilibrium
- Q > K: rxn shifts leftwards, towards reactants (too much P)
- Q < K: rxn shifts rightwards, towards products (too much R)
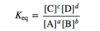
Application of Le Chatelier’s Principle
- Change in concentration, pressure, or temperature causes a system at equilibrium to shift in a direction to reduce the stress
- Change in concentration—-> rxn shifts in direction of less concentrated side
- Increase in pressure—->rxn shifts towards side of rxn with less moles of gas
- Increase in temperature—–>rxn shifts away from the side of the equation with more heat (towards gaseous side)
Relationship of the equilibrium constant and ΔGo
What is the equation?
- If Keq = 1, then ΔGo =?
- If Keq > 1, then ΔGo = ?
- If Keq < 1, the n ΔGo = ?
- To determine if a reaction is spontaneous under specific conditions (not just standard conditions), both Q (concentrations of reactant and product) and ΔG must be considered
o ΔG = ΔGo + RTln(Q)
* ****Can be rewritten as ΔG<sup>o</sup>= -RTLin (K) * **If Keq = 1,** then ΔG<sup>o</sup> = 0 * **If Keq \> 1**, then ΔG<sup>o</sup> \< 0 * **If Keq \< 1,** the n ΔG<sup>o</sup> \> 0 * This only determines **spontaneity at a specific temperature** (Keq and ΔG<sup>o</sup> values depend on **_TEMPERATURE)_**
