1 - 100 Flashcards
A 16 month-old girl presents with patchy alopecia, whorled erythematous scaly eruption, and asymmetric limb shortening. What laboratory or radiologic test may aid in diagnosis?
1 Brain MRI
2 Alkaline phosphatase
3 Chest radiograph
4 Bone films
5 Complete blood count
Bone films
The patient has Conradi-Hunermann Syndrome. This is a X-linked dominant disorder characterized by ichthyosiform erythroderma in Blaschko’s lines in infancy which resolves with follicular atrophoderma, patchy alopecia, short stature, cataracts, scoliosis, assymetric limb shortening. Bone films will demonstrate stippled epiphyses. Ichthyosis and stippled epiphyses resolve after infancy.
A patient with 20 nail dystrophy, steatocystoma multiplex and natal teeth likely has a mutation in the genes coding for:
1 Keratins 5
2 Laminin 5
3 Plakophilin 1
4 Keratins 6b & 17
5 Keratins 6 &16
Keratins 6b & 17
Pachyonychia congenital is an autosomal dominant condition with 20 nail dystrophy. The patient described has Type II (Jackson-Sertole) disease, which includes steatocystoma multiplex, natal teeth, multiple cysts, and micropthalmia, and is caused by mutations in keratins 6b& 17. Type I (Jadassohn-Lewandowsky) also includes focal symmetric PPK, follicular hyperkeratosis, oral leukokeratoses and is caused by mutations in keratins 6 &16. Type III includes the clinical features of type I + corneal leukokeratosis. Mutations in keratins 5&14 represents EB simplex, Laminin 5 mutation is seen in Junctional EB, and plakophilin 1 mutation is seen in ectodermal dysplasia with skin fragility.
Which of the following is defective in Ehlers-Danlos syndrome (EDS) with congenital adrenal hyperplasia?
1 Tenascin-X
2 Lysyl oxidase
3 Lysyl hydroxylase
4 None of these answers are correct
5 All of these answers are correct
Tenascin-X
Tenascin-X defects are associated with EDS and with congenital adrenal hyperplasia. The phenotype is that of typical EDS with hyperextensible skin, hypermobile joints, and tissue fragility. Lysyl oxidase is defective in X-linked EDS (type V) and Occipital horn syndrome (type IX). Lysyl hydroxylase is defective in ocular-scoliotic (type VI) EDS.
Which type of porphyria is associated with hyponatremia?
1 Acute intermittent porphyria
2 Porphyria cutanea tarda
3 Variegate porphyria
4 Hereditary coproporphyria
5 Erythropoietic protoporphyria
Acute intermittent porphyria
Acute intermittent porphyria can cause hyponatremia due to the syndrome of inappropriate antidiuretic hormone secretion.
Hypoplasia of the breast can be seen in which disease?
1 Anhidrotic ectodermal dysplasia
2 Maffucci syndrome
3 Congenital syphilis
4 Marfan syndrome
5 Osteogenesis imperfecta
Anhidrotic ectodermal dysplasia
Anhidrotic ectodermal dysplasia is a X-linked recessive disease caused by mutations in ectodysplasin, a member of the tumor necrosis family. Patients may have dry skin with pigmentation periorbitally, hypohidrosis, sparse hair, hypo-anodontia, nail dystrophy, and frontal bossing, and saddle nose deformity. In addition to abnormalities of other ectodermally derived structures, the breast and nipple-areolar complex may be absent or hypoplastic.
Which disease is found more commonly in mothers of patients with chronic granulomatous disease?
1 Sarcoidosis
2 Erythema nodosum
3 Churg-Straus disease
4 Wegener’s disease
5 Discoid lupus erythematous
Discoid lupus erythematous
Female carriers of chronic granulomatous disease have an increase incidence of discoid lupus, infections and apthous stomatitis.
A patient presents with several light blue cyst-like lesions on the eyelid. They consult their list of problems and bring up plantar hyperkeratosis and dysplastic toenails. On oral exam, you note that they have both upper and lower dentures. The patient relates that after losing their “baby teeth”, only 3 teeth grew in their place. What syndrome does this person most likely have?
1 Schopf-Schulz-Passarge
2 Gardner syndrome
3 Hypohidrotic ectodermal dysplasia
4 Cowden syndrome
5 Cronkhite-Canada
Schopf-Schulz-Passarge
Schopf-Schulz-Passarge syndrome is associated with hydrocystomas of the eyelids, hypotrichosis (near complete loss of hair early in life), hypodontia, nail abnormalities and multiple palmoplantar eccrine syringofibroadenomas. The other listed syndromes do not fit the description above.
A child presenting with the scalp findings shown and a right arm hypoplasia would be diagnosed with which of the following syndromes?
1 Adams-Oliver syndrome
2 Bart’s syndrome
3 Progeria
4 Dunnigan syndrome
5 None of these options are correct
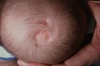
Adams-Oliver syndrome
Adams-Oliver syndrome is defined by aplasia cutis congenita (ACC) (shown in the image), usually of the midline scalp with limb hypoplasia. Bart’s syndrome also has ACC as a finding, but it is usually present on the lower extremities and associated dominant dystrophic epidermolysis bullosa. Progeria is a premature aging syndrome associated with a lamin-A mutation. Dunnigan syndrome is also known as Familial partial lipodystrophy and is associated with a mutation in the BSCL2 gene. Neither are associated with findings of ACC.
Which of the following findings is characteristic of a mutation in lamin A?
1 Lipoatrophic sclerodermoid skin
2 Alopecia
3 Craniomegaly with small face
4 Severe premature atherosclerosis with early death
5 All of the answers are correct
All of the answers are correct
A mutation in Lamin A causes Progeria (Hutchinson-Gilford syndrome). Other findings include nail atrophy and muscle/bone wasting. Presentation is in the first or second year of life. An increased urine hyaluronic acid can be helpful in diagnosis.
What is the mode of transmission for lamellar ichthyosis?
1 Autosomal dominant
2 Autosomal recessive
3 X-linked dominant
4 X-linked recessive
5 Sporadic

Autosomal recessive
Lamellar ichthyosis which is characterized by collodian membrane in newborns and platelike scale in children and adults is an autosomal recessive syndrome. The gene defect is transglutaminase 1 (TGM1).
Think Transglutaminase Fetal Rat
The most common cutaneous association with monilethrix is:
1 Eczema
2 Hypopigmentation
3 Hyperpigmentation
4 Keratosis Pilaris
5 Atrophy

Keratosis Pilaris
Monilethrix is an autosomal dominant condition which, by definition, presents with “beaded” hear. Clinically, patients present with short, sparse lusterless hair. Keratosis pilaris is the most common associated feature.

Which type of epidermolysis bullosa simplex is associated with early death?
1 Weber-Cockayne
2 Generalized (Koebner)
3 Dowling-Maera
4 Ogna variant
5 Non-Herlitz variant
Dowling-Maera
The Dowling-Maera variant of epidermolysis bullosa simplex is associated with widespread bullae, significant mucous membrane and laryngeal/esophageal involvement, nail dystrophy, and early death.
The arylsulfatase C gene is mutated in which disease?
1 X-linked ichthyosis
2 Refsum syndrome
3 Haim-Munk syndrome
4 Naxos syndrome
5 Griscelli syndrome
X-linked ichthyosis
Arylsulfatase C is also known as steroid sulfatase and is mutated in X-linked ichthyosis. This condition is inherited in a X-linked recessive pattern. Clinical findings include: brown scale sparing palms, soles and flexures, comma-shaped corneal opacities, failure of labor progression and cryptorchidism. It is also mutated in X-linked recessive type chondrodysplasia punctata.
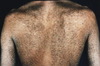
Premalignant leukoplakia of the oral mucosa is associated with:
1 Bloom syndrome
2 Werner Syndrome
3 Xeroderma Pigmentosum
4 Dyskeratosis Congenita
5 Rothmund-Thomson syndrome
Q/Q(M)-474043 Report a Problem

Dyskeratosis Congenita Dyskeratosis Congenita (also known as Zinsser-Engman-Cole syndrome) is thought to have two modes of inheritance. The more common X-linked disorder is due to a mutation in the Dyskerin gene, while the autosomal dominant form is due to a mutation in TERC, a telomerase RNA component. Clinical features include reticulated gray-brown hyperpigmentation, paloplantar hyperkeratosis, alopecia, onychodystrophy, premalignant leukoplakia of any mucosal surface, and mental retardation.

Which cutaneous finding is seen in patients with phenylketonuria?
1 Angular stomatitis
2 Ichthyosis
3 Pigment dilution of hair and skin
4 Phyrnoderma
5 Erosive diaper dermatitis
What is the enzyme defect?
The accumulation of ally
What should the PKU patients avoid eating in their diet?

Pigment dilution of hair and skin
Phenylketonuria is an autosomal RECESSIVE condition caused by a mutation in the gene coding for phenylalanine hydroxylase**. Defect in this enzyme results in accumulation of phenylalanine and its metabolites. I_ncreased phenylalanine_** has toxic effects on the central nervous system in addition to competitively inhibiting tyrosine in melanogenesis. Inhibition of melanogenesis results in pigmentary dilution of the hair and skin. Other features of this condition include a predisposition to eczema, sclerodermoid changes of the skin, urine that has a distinctive “mousy” odor, psychomotor delay, mental retardation, seizures and hyperreflexia. A low-phenylalanine diet instituted early on can prevent these manifestations of the disease. The morbidity of phenylketonuria has improved since the advent of routine neonatal screening for this condition.
Phenylalanine ——-> Tyrosine —-> Melanin
+Phenylanine Hydroxylase
Phenyl Ketonuria (PKU) is a common metabolic disorder in growing children. A child suffering from PKU has defective legs and is usually unable to stand. Such a child may also be mentally retarded. This condition is due to a defect in the amino acid metabolism. In the course of amino acid metabolism there is a step in which the amino acid phenyl-alanine gets converted into another amino acid called tyrosine. This conversion requires specific enzyme called phenyl pyruvic hydroxylase. In the child suffering from PKU this particular enzyme will be absent and as a result phenyl-alanine gets converted into a ketone body phenyl pyruvic acid. It starts circulating throughout the body through the blood resulting in the disorder. The absence of the enzyme phenyl pyruvic hydroxylase is due to the expression of a recessive autosomal gene in the homozygous condition.
PKU can be detected by a simple urine test. Such children are usually treated with a diet, which is free from the amino acid phenyl-alanine.
Which syndrome is characterized by hyperhidrosis, lack of pain sensation, hypersalivation, and absent fungiform papillae?
1 Turner Syndrome
2 Noonan Syndrome
3 Riley-Day
4 Rubinstein-Taybi syndrome
5 Cornelia de lange Syndrome

Riley-Day
“Riley Day the IRON CHEF cutting his fingers”
Riley-Day syndrome is also known as Familial Dysautonomia. It is an autosomal RECESSIVE disorder with the gene defect on the long arm of chromosome 9.
Patients have unmyelinated sensory and sympathetic neurons and autonomic dysfunction, leading to hyperhidrosis, decreased corneal sensation and tear flow, hypersalivation, gastroesophageal reflux, decreased deep tendon reflexes, and lack of pain sensation.
They also exhibit abnormal histamine skin test.
Retention of primary teeth a dental finding of which of the following conditions?
1 Hypomelanosis of Ito
2 Letterer-Siwe disease
3 Tuberous sclerosis
4 Jackson Sertoli syndrome
5 Hyper-IgE syndrome

Hyper-IgE syndrome
Hyper-Immunoglobulin E syndrome is an autosomal dominant condition with impaired regulation of IgE function and deficient neutrophil chemotaxis. There is increased susceptibilty to infections and increased IgE serum levels. Retained primary teeth and lack of development of secondary teeth are characteristic findings. The remaining conditions do not have this as a prominent finding.

All of the following disorders are exacerbated by UV radiation and have the eye findings in photo except:
1 Bloom syndrome
2 Hartnup’s disease
3 Refsum syndrome
4 Cockayne syndrome
5 Rothmund-Thomopson syndrome

Refsum syndrome
Refsum’s syndrome is an autosomal recessive disorder caused by mutations in phytanoyl-CoA hydroxylase. Clinically, patients have mild icthyosis, cerebellar ataxia, polyneuropathy, salt and pepper retinitis pigmentosa, sensorineural deafness, and arrhythmias with heart block. They are not overly sensitive to UV radiation.

Homocystinuria is caused by a defect in:
1 Phenylalanine hydroxylase
2 Biotinidase
3 Holocarboxylase synthetase
4 Cystathione beta-synthetase
5 Gp91-phox
What is the most common eye abnormailty?
Cystathione beta-synthetase
Cystathione beta-synthetase is defective in homocystinuria, an autosomal recessive conditions characterized by increased homocystine and methionine levels in blood and urine. Other findings include a malar flush, DVTs/emboli, cardiovascular disease, livedo reticularis, leg ulcers, blonde hair/fiar complexion, downward lens dislocation, glaucoma, mental retardation, seizures, psychiatric disorders and a marfanoid body habitus. The other enzymes are not involved in this condition.
Cutaneous osteomas are seen in which syndrome?
1 Waardenburg syndrome
2 LEOPARD syndrome
3 Carney complex
4 Albright hereditary osteodystrophy
5 Gaucher�s syndrome
Albright hereditary osteodystrophy is caused by mutations in the Gs subunit of adenylate cyclase. There is calcification and ossification due to pseudohypoparathyroidism, absent 4th knuckle, and hypogonadism.
Q/Q(M)-474257 Report a Problem
A child with phenylketonuria likely presents with which cutaneous problems?
1 Blue-gray generalized hyperpigmentation
2 Alopecia universalis
3 Generalized hypopigmentation
4 Generalized hyperpigmentation
5 Leg ulcers
Phenylketonuria is an autsomal recessive disorder caused by a mutation on the long arm of chromosome 12. A deficiency of phenylalanine hydroxylase or its cofactor tetrahydrobiopterin leads to accumulation of phenylalanine. Clinical features include generalized hypopigmentation, eczematous dermatitis, sclerodermoid changes, seizures, psychomotor delay, urine with �mousy� odor, mental retardation.
Q/Q(M)-474030 Report a Problem
Patients with junctional epidermolysis bullosa have been found to have mutations in:
1 Laminin 5
2 Bullous pemphigoid antigen 2
3 Collagen 17
4 BP180
5 All of the answers are correct
All of the answers are correct. Laminin 5 is a protein integral in the adhesion of the dermis to the epidermis. Also involved in junctional epidermolysis bullosa is bullous pemphigoid antigen 2, collagen 17 and BP180, which are synonymous for the same structure.
Q/Q(M)-477712 Report a Problem
Which genetic defect could explain cutaneous findings in addition to abnormal immunoglobulin levels, recurrent respiratory infections, hypogonadism, and an increased risk of leukemia and lymphoma?
1 RecQL3
2 ERCC6
3 WAS gene
4 NADPH oxidase
5 Adenosine deaminase

Bloom’s syndrome is an autosomal recessive disorder caused by mutations in the RecQL3 gene encoding a DNA helicase. Clinically, individuals with Bloom’s syndrome have a photodistributed erythema with telangectasia on the malar eminences.
The may also have decreased IgM and IgA levels, hypogonadism**, and an increased risk for **leukemia and lymphoma.

What is the underlying gene defect for this transgrediens form of palmoplanter keratoderma
1 SLURP-1
2 TOC gene
3 Plakoglobin
4 Keratin type 1
5 Keratin type 9

Attached picture is Mal de Meleda (keratosis palmoplantaris transgrediens) which is an autosomal recessive form of diffuse PPK, associated with keratotic plaques that extend to the dorsal aspects of the hands and feet (“transgrediens”) and may overlie joints . Hyperhidrosis, superinfection, and occasionally perioral erythema, brachydactyly, and nail abnormalities are associated. Mal de Meleda is due to mutations in ARSB, which encodes SLURP-1. The other choices represent gene defects for “non-transgrediens” forms of PPK (Plakoglobin in Naxos syndrome, TOC gene in Howel-Evans syndrome, K1 in non-epidermolytic PPK “Unna-Thost”, and K9 in epidermolytic PPK “Vorner”

A child presents with pretibial hyperpigmentation, ataxia, decreased motor coordination, cirrhosis, and decreased motor coordination. The physical exam which would reveal the most specific finding for this disease is:
1 Hearing test
2 Slit-lamp eye exam
3 EKG
4 Colonoscopy
5 Renal ultrasound
Wilson’s disease (also known as hepatolenticular degeneration) is an autosomal recessive disorder result in defective biliary excretion of copper, leading to copper accumulation in the liver, brain and cornea. Clinical features include hepatomegaly, cirrhosis, ataxia, dysarthria, decreased motor coordination, pretibial hyperpigmentation, blue lunulae, and copper deposition in the cornea�Kayser-Fleisher ring, which can be diagnosed using a slit-lamp.
Q/Q(M)-474029 Report a Problem
Nevoid basal carcinoma syndrome (Gorlin syndrome) is autosomal dominant transmitted mutation of the patched gene. Symptoms include innumberable basal cell carcinomas, painful odontogenic jaw keratocysts, palmoplantar pits, frontal bossing, bifid ribs and what other bony abnormality?
1 Polyostotic fibrous dysplasia
2 Stippled epiphyses
3 Calcification of falx cerebri
4 Osteopoikilosis
5 Sphenoid wing dysplasia
Calcification of falx cerebri is seen in Gorlin’s syndrome. CHILD syndrome and chondrodysplasia punctata both can exhibit stippled epiphyses. Polyostotic fibrous dysplasia is found in McCune-Albright syndrome, osteopoikilosis in seen in Buschke-Ollendorf syndrome. Sphenoid wing dysplasia is seen in neurofibromatosis type I.
A Puerto Rican woman is seen in clinic for a pruritic rash on her trunk. A punch biopsy is performed. The biopsy site continues to bleed, with hematoma formation. The bleeding is eventually controlled. On further exam, her skin and hair are light brown. She has a history of granulomatous colitis. What it the most likely reason she had excess bleeding with a simple procedure?
1 Her platelets lack dense bodies, causing excess bleeding
2 Her intrinsic factor is deficient
3 Her Factor VIII levels are low
4 She is congentially deficient in platelets
5 None of the answers are correct
Platelets without dense bodies cause excess bleeding in Hermansky-Pudlak syndrome. Other features of this condition include oculocutaneous albinism, ceroid lysosomal storage disease resulting in pulmonary fibrosis, granulomatous colitis.
A patient with port wine stain on a lower extremity, hemihypertrophy of the limb and lymphatic and deep venouse insufficiency of the affected limb would be considered to have Klippel-Trenaunay-Weber syndrome. What additional feature would need to be present to define the patient as having Parkes-Weber syndrome?
1 Arteriovenous fistulas
2 Multiple cafe-au-lait macules
3 Macroglossia
4 Cutis marmorata
5 Distichiasis
Parkes-Weber syndrome has the additional feature of arteriovenous fistulas. The remaining features are not part of these syndromes.
Which of the following diseases is caused by a mutation in a gap junction protein?
1 Striated PPK
2 Schopf-Schulz-Passarge syndrome
3 Mal de Meleda
4 Vohwinkel syndrome (classic)
5 Vohwinkel syndrome (ichthyotic)
Classic Vohwinkel syndrome is caused by mutations in connexin 26, a gap junction protein. Ichthyotic Vohwinkel syndrome is caused by mutations in loricrin and has ichthyosis but not deafness.
A patient with Bloom Syndrome is most likely to have which laboratory abnormalities:
1 Decreased immunoglobulins
2 Macrocytic anemia
3 Elevated IgE
4 Thrombocytopenia
5 Positive ANA
Decreased immunoglobulins
Bloom syndrome is an autosomal recessive disorder due to a mutation in the BLM gene which codes for a DNA helicase. Patients have impaired DNA repair after UV exposure and increased photosensitivity. Clinical features include photodistributed erythema, cheilitis, high-pitched voice, hypogonadism, and increased risk for leukemia, lymphoma and GI adenocarcinoma. Laboratory evaluation reveals decreased IgA, IgM and IgG leading to increased risk of respiratory infections
Which of the following features is not associated with Cornelia de Lange Syndrome?
1 Normal intelligence
2 Characteristic facies with downturned mouth, hirsutism, synophrys, trichomegaly, anteverted nostrils, long philtrum and low set ears
3 Cryptorchidism
4 Fifth finger clinodactyly
5 Recurrent lung infections
Normal intelligence
Children with Cornelia de Lange are usually severly retarded with an IQ <35. In addition to the features listed above, other features include cutis marmorata, hypoplastic nipples and umbilicus, low-pitched cry in infancy and congenital heart defects. While most cases are inherited in a sporadic manner, those cases which are familial are thought to be autosomal dominant and associated with the NIPBL (nipped-beta-like) gene. Prognosis is poor with premature death often secondary to sspiration or recurrent pulmonary infection.
Defects in Fibrillin 2 are linked with:
1 Congenital contractural arachnodactyly
2 Cutis Laxa
3 Arthrochalasis multiplex congenita
4 Occipital horn syndrome
5 Lipoid proteinosis
Fibrillin 2 defects arelinked primarily with congenital contractural arachnodactyly. This syndrome is associated with long limbs, arachnodactyly, scoliosis and crumpled ears. Occasionally, fibrillin 2 can be associated with Marfan syndrome also. The other conditions are not linked to fibrillin mutations.
The finding of ‘maltese crosses’ in the urine is characteristic of which of the following conditions?
1 Alkaptonuria
2 Fabry disease
3 Gaucher disease
4 Neimann-Pick disease
5 Hunter syndrome
The ‘maltese cross’ finding in urine is characteristic of Fabry disease. Alkaptonuria will show dark urine with a pH > 7.0. There are no urinary findings in Hunter syndrome, Gaucher or Neimann-Pick disease.
A 12 year-old boy with pits on his palms and lateral fingers may have:
1 Arsenic exposure
2 A hereditary keratoderma
3 A corynebacteria infection
4 An inherited cancer syndrome
5 Secondary syphilis
Basal cell nevus syndrome is an autosomal dominant disease caused by mutations in the PTCH1 gene. Clinically, patients may have numerous basal cell carcinomas, palmoplantar pits, jaw cysts, frontal bossing, bifid ribs, calcification of falx cerebri, medulloblastoma, ovarian fibromas and fibrosarcomas.
The characteristic dental findings in patients with tuberous sclerosis are:
1 Peg teeth
2 Anodontia
3 Enamel pits
4 Odontogenic cysts
5 Retention of primary teeth
Enamel pits are the characteristic dental findings in tuberous sclerosis. Peg teeth are found in incontinentia pigmenti and anhidrotic ectodermal dysplasia. Anodontia is found in hypomelanosis of ito and incontinentia pigmenti. Odontogenic cysts are seen in Gorlin syndrome, and retention of primary teeth is characteristic of Job syndrome.
What is the classic radiologic findings associated with this disorder?
1 Dural calcifications
2 Calcifications of the falx-cerebri
3 Tram-track calcifications of the temporal and occipital cortex
4 Osteopatha striata
5 Osteopoikilosis

Sturge-Weber syndrome is a sporadic disroder characterized by a facial capillary malformation in a trigeminal nerve distribution. Patients with Sturge-Weber may have cerebral atrophy, ipsilateral vascular malformations of the leptomeninges, seizures, and glaucoma. The classic radiologic finding is tram-track calcifications of the temporal and occipital cortex.
On cutaneous exam, angiokeratoma corporis diffusum is characteristic of which of the following conditions?
1 Sialodosis
2 Fucosidosis
3 Fabry disease
4 All of these options are correct
5 None of these options are correct
Findings of angiokeratoma corporis diffusum are found in all three listed conditions. They cannot by distinguished by skin exam.
Painful crises and ‘whorled’ corneal opacities are seen with which of the following enzyme abnormalities?
1 Homogentisic acid oxidase
2 Alpha-galactosidase A
3 Glucocerebrosidase
4 Iduronate sulfatase
5 Glucoronidase
Painful crises and whorled corneal opacities are found in Fabry disease which is caused by a defect in alpha-galactosidase A. The remaining conditions do not have these findings.
Tyrosinase positive albinism (oculocutaneous albinism type 2) is caused by a mutation in which of the following:
1 Tyrosinase
2 P gene
3 Tyrosinase related protein 1
4 C-kit
5 NEMO
P gene
Oculocutaneous albinism (OCA) type 1 (Tyrosinase negative albinism) is caused by mutations in the tyrosinase gene. OCA type 2 (tyrosinase positive albinism) is caused by mutations in the P gene. OCA type 3 is caused by mutations in the tyrosinase related protein 1 gene. C-kit mutations cause piebaldism and NEMO mutations cause incontinentia pigmenti.
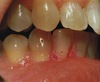
Underlying defect for the disease shown in picture is
1 ATP2A2
2 ATP2C1
3 BPAG1
4 BPAG2
5 Collagen type 17

ATP2C1
The disease shown in image is Hailey-Hailey disease (Familial Benign Pemphigus) which is an autosomal dominant genodermatosis, caused by mutation in ATP2C1, encoding a calcium pump protein related to SERCA2. It is characterized by recurrent vesicles and erosions, which most commonly appear on the sides and back of the neck, in the axillae, in the groin, and in the perianal regions. The disorder is not seen before puberty and usually has its onset in the late teens or early 20s. In the intertriginous area lesions tend to form erythematous plaques with dry crusting and soft, flat, and moist granular vegetations. Burning or pruritus is common, and, particularly in the intertriginous areas, lesions tend to become irritating, painful, and exceedingly uncomfortable. ATP2A2 is underlying defect in Darier’s disease, other choices are defects seen in pemphigoid and epidermolysis bullosa.
Q/Q(M)-482120 Report a Problem
Which opthamologic disease is associated with this disorder?
1 Glaucoma
2 Ectopia lentis
3 Cataracts
4 Posterior subcasular lentiular opacity
5 Retinitis pigmentosa

Glaucoma
Sturge-Weber syndrome is a sporadic disease characterized by a capillary malformation in the trigeminal distribution. Patients may have associated cerebral atrophy, vascular malformations of the leptomeninges, and seizures. All patients with Sturge-Weber should be referred to the opthamologist for glaucoma screening.

The presence of natal teeth and pincer nails suggests which disease entity?
1 Congenital syphillis
2 Thalidomide exposure in utero
3 Incontinentia pigmenti
4 Pachyonychia congenita
5 Anhidrotic ectodermal dysplasia

Pachyonychia congentia is an autosomal dominant condition characterized by a constellation of findings affecting ectodermal structures. These include the presence of natal teeth, steatocystoma multiplex, follicular hyperkeratosis of the knees, elbows and extensor extremities, eruptive vellus hair cysts, and oral leukokeratosis which is not pre-malignant. In addition, nail findings include twenty-nail dystrophy, subungual hyperkeratosis with increase transverse curvature (“pincer nails”) and candidal paronychia. There are two forms of pachyonychia congenital: Type 1 (Jadassohn-Lewandowsky syndrome) caused by defects in keratin 6a and 16, and Type 2 (Jackson-Lawler type) caused by defects in keratins 6b and 17. Anhidrotic ectodermal dysplasia is associated with peg-shaped teeth, hypoanodontia, and a non-specific nail dystrophy. Likewise, incontientia pigmenti also is characterized by anodontia and peg-shaped teeth and dystrophic changes of the nail. Finally congenital syphilis is a well-recognized cause of pegged teeth. Limb deformities are the most serious sequelae of thalidomide exposure in utero.

What is the gene defect in harlequin fetus?
1 Transglutaminase
2 Steroid sulfatse
3 ABCA12
4 ABCC6
5 None of these answers are correct

Harlequin fetus is an autosomal recessive disorder. The gene defect is ABCA12.
Spastic ditetraplegia is associated with which of the following disorders?
1 Sjogren-Larsson syndrome
2 X-linked ichthyosis
3 Lamellar ichthyosis
4 KID syndrome
5 Refsum syndrome

Sjogren-Larsson syndrome is an autosomal recessive disorder caused by mutations in the fatty aldehyde oxidoreductase/alcohol dehydrogenase gene. This disorder is characterized by ichthyosis, spastic ditetraplegia, mental retardation, epilepsy, glistening dot retinal pigmentation, and dental enamel dysplasia.

What protein is deficient in the condition shown?
1 Calcium ATP�ase IIC1
2 Calcium ATP�ase IIA2
3 PEX-7
4 SPINK5
5 Desmoglein 3

The picture shown is Hailey-Hailey disease. This is an autosomally dominant condition with a defect in Calcium ATP�ase IIC1. On H&E stain, an acantholytic �dilapidated brick wall� appearance is seen. Calcium ATP�ase IIA2 is defective in Darier�s Disease, PEX-7 in autosomal recessive type Conradi-Hunermann disease, SPINK5 in Netherton�s disease and Desmoglein 3 in pemphigus vulgaris.
A patient Buschke-Ollendorff syndrome has osteopoikilosis and which cutaneous finding?
1 Waxy papules along the eyelids
2 Caf� au lait macules
3 Port wine stain
4 Juvenile elastoma
5 Epidermal nevi
Juvenile elastoma
Buschke-Ollendorf syndrome is an autosomal dominant syndrome associated with increased elastic fiber in the skin. Key features include dermatofibrosis lenticularis disseminata (also called juvenile elastomas) and osteopoikilosis.

Which of the following metals is deficient in the serum of patients with Menkes kinky hair syndrome?
1 Copper
2 Iron
3 Selenium
4 Zinc
5 Biotin
Menkes kinky hair syndrome is transmitted in an X-linked recessive manner and is caused by a mutation in ATP7A, an ATP-dependent copper tranporter. This defect results in low serum levels of copper. These individuals will have hair abnormalities such as sparse, hypopigmented brittle hair, eyelashes and eyebrows, lax skin, a “cupid’s bow” upper lip, CNS progressive deterioration, seizures, skeletal abnormalities and tortuous arteries. The other listed items are not associated with Menkes syndrome.

Which syndrome is characterized by broad thumbs, a large beaked nose, and capillary malformation?
1 Klinefelter
2 Proteus syndrome
3 Bloom syndrome
4 Rubinstein-Taybi
5 Ehlers-Danlos syndrome

Rubinstein-Taybi syndrome has been associated with a deletion localized to the short arm of chromosome 16. Patients are severely retarded with strabismus, crytorchidism, and congenital heart defects. They have a characteristic beaked nose with nasal septum below alae accompanied by a broad nasal bridge, downslanting palpebral fissures, and broad thumbs and halluces.

What medication may exacerbate this autosomally dominant, acnatholytic disorder?
1 Phenytoin
2 Lithium
3 Oral contraceptives
4 Anti-malarials
5 Corticosteroids
Lithium
Darier’s disease is autosomal dominant condition characterized by hyperkeratotic papules coalescing into warty plaques and cobblestoned papules on mucosal surfaces. The cutaneous manifestations may be exacerbated by lithium.

Anodontia is a bone finding seen in which of the following conditions:
1 Hypomelanosis of Ito
2 Letterer-Siwe disease
3 Tuberous sclerosis
4 Jackson Sertoli syndrome
5 Hyper-IgE syndrome
Hypomelanosis of Ito, or Incontinentia pigmenti achromians is a condition characterized by marble-cake hypopigmentation, epilepsy, alopecia, scoliosis and mental/motor retardation. The characteristic dental abnormality is anodontia. The remaining syndromes are not associated with anodontia.

In patients with diffuse congenital hemangiomatosis, the most common site for extracutaneous involvement is the :
1 Liver
2 Thyroid
3 Lungs
4 Colon
5 Brain
Diffuse congenital hemangiomatosis is characterized by multiple hemangiomas with the liver being the most common extracutaneous site, followed by the lungs. Liver hemangioma may be complicated by hepatomegaly, obstructive jaundice, and portal hypertension.

You are consulted on a patient with possible Nethertons Syndrome. Which location of the body would most likely have hairs demonstrating trichorrhexis invaginata?
1 Scalp
2 Eyebrow
3 Eyelash
4 All of these answers are correct
5 None of these answers are correct
Eyebrow hair is most common site with hairs demonstrating trichorrhexis invaginata.

Dermatofibrosis lenticularis disseminata is seen in which of the following conditions?
1 Ehlers-Danlos syndrome
2 Marfan syndrome
3 Pseudoxanthoma elasticum
4 Focal dermal hypoplasia
5 Buschke-Ollendorf syndrome
Buschke-Ollendorf syndrome is an autosomal dominant disorder characterized by dermatofibrosis lenticularis disseminata (cutaneous elastomas distributed symmetrically over the buttocks, trunk and proximal extremities), and osteopoikilosis (round opacities in bones).

An infant presents with multiple congenital hemangomas in an generalized distribution. What is the most serious associated condition?
1 Congestive Heart Failure
2 Obstructive jaundice
3 Portal hypertension
4 All of the answers are correct
5 None of the answers are correct
Congestive Heart Failure
High output congestive heart failure can lead to death in these children. Obstructive jaundice and portal hypertension both occur, but are less likely to cause death. The hemangiomas will undergo spontaneous regression.

Patients with Chondrodysplasia punctata can have findings of stippled epiphyses on X-ray examination. Which other x-linked dominant condition can have stippled epiphyses?
1 CHILD syndrome
2 Incontinentia Pigmenti
3 Focal Dermal Hypoplasia
4 Goltz syndrome
5 Bazex syndrome
CHILD syndrome
All of the syndromes listed have X-linked dominant inheritance. CHILD syndome also has findings of stippled epiphyses. Incontinentia pigmenti is caused by defecdts in the NEMO gene. Findings include peg/conical teeth, eye and CNS defects and alopecia. There are no bone abnormalities. Focal Dermal Hypoplasia, otherwise known as Goltz syndrome has findings of linear atrophy following Blaschko’s lines with areas of fat herniation, mucocutaneous papillomas and pits, alopecia, nail dystrophy, tooth abnormalities and osteopathia striata (striations of the long bones). Bazex syndrome is associated with follicular atrophoderma, hypohidrosis, hypotrichosis and multiple basal cell carcinomas. There are no bone abnormalities associated.

Findings of milia, cylindromas and the condition shown in the pathology image are characteristic of which of the following syndromes?
1 Gorlin’s syndrome
2 Familial cylindromatosis
3 Brook-Spiegler syndrome
4 Rasmusen syndrome
5 Rombo syndrome

Brook-Spiegler syndrome
Brooke-Spiegler syndrome is an uncommon disease with a predisposition to develop cutaneous adnexal neoplasms such as cylindromas, trichoepitheliomas, spiradenomas, trichoblastomas, basal-cell carcinomas, follicular cysts, organoid nevi, and malignant transformation of pre-existing tumors in the affected individuals.

What nail change is seen in patients with Mal de Meleda Syndrome?
1 Onycholysis
2 Longitudinal ridging
3 Koilonychia
4 leukonychia
5 pterygium
Koilonychia
Mal de Meleda is an autosomal recessive disease characterized by transgedient malodorous PPK, hyperhidrosis, keratotic plaques at knees and elbows, subungual hyperkeratosis, and koilonychia. The gene defect is SLURP 1.
Meleda Puzzle SLURP-1

The most common cardiovascular defect in patients with Noonan syndrome is:
1 Atrial septal defect
2 Ventricular septal defect
3 Enlarged aorta
4 Pulmonic valve stenosis
5 Aortic stenosis
Noonan’s syndrome is also known as cardiofaciocutaneous syndrome. Patients have short stature, ptosis, hypertelorism, low-set ears, thick lips and curly hair. Pulmonic valve stenosis is the most common cardiovascular defect, with atrial septal defects also seen.
It is type of dwarfism, a genetic disorder (hereditary) that prevents normal development or causes abnormal development of various parts of the body. Referred as an autosomal dominant condition, this defect affects genes; which are responsible for individual’s growth.Some symptoms of the disease are mild mental retardation, unusual chest shape**, delayed puberty and **short neck. Current 7 genes are known that can cause this disorder and the main cause is mutation in those genes. There is no specific cure known for this disease. Treatment depends upon the stage and type of the disease. Growth hormones can be way out for the short stature.
Hypertelorism, derived from the Greek word “telouros” (meaning distant), is an abnormally increased distance between two organs or bodily parts, usually referring to an increased distance between the orbits (eyes)–orbital hypertelorism. In this condition the distance between the inner eye corners as well as the distance between the pupils is greater than normal. Hypertelorism should not be confused with telecanthus, in which the distance between the inner eye corners is increased but that of the outer eye corners remains unchanged.[1] Therefore the distance between the pupils is normal.

Dermatofibrosis lenticularis disseminata and osteopoikilosis are findings seen with mutations of which of the following genes?
1 LEMD3
2 Fibrillin 2
3 ABCC6
4 Lysyl hydroxylase
5 Lysyl oxidase
What stain will confirm the diagnosis of dermatofibrosis lenticularis disseminata?

Buschke-Ollendorf syndrome is caused by a loss-of-function mutation in LEMD3.
Stains with Elastic fibers = Verhoeff-van Gieson
Dermatofibrosis lenticularis disseminata are small papules or discs of increased dermal elastic tissue appearing in early life; when osteopoikilosis is also present, the condition is called osteodermatopoikilosis or Buschke-Ollendorf syndrome; autosomal dominant inheritance. (05 Mar 2000)
The radiographic appearance of osteopoikilosis on an x-ray is characterized by a pattern of numerous white densities of similar size spread throughout all the bones. This is a systemic condition. It must be differentiated from blastic metastasis, which can also present radiographically as white densities interspersed throughout bone. Blastic metastasis tends to present with larger and more irregular densities in less of a uniform pattern. Another differentiating factor is age, with blastic metastasis mostly affecting older people, and osteopoikilosis being found in people 20 years of age and younger.

Which malignancy is seen in approximately 15-20% of people with the disease characterized by a defect in a parathyroid hormone receptor protein?
1 Osteosarcoma
2 Angiosarcoma
3 Chondrosarcoma
4 Rhabdomyosarcoma
5 Epitheliod sarcoma
Approximately 15-20% of patients with Maffucci’s syndrome will develop chondrosarcoma. Maffucci’s syndrome is due to a defect in a parathyroid hormone receptor protein.

Ivory-colored papules between the angles of the scapulae are characteristic of which syndrome:
1 Hurler
2 Scheie
3 Morquio
4 Hunter
5 Sanfilippo
These syndromes are all mucopolysaccharidoses. These papules are characteristic of Hunter syndrome which is caused by a deficiency in iduronate sulfatase.
Hunter syndrome, or mucopolysaccharidosis Type II, is a lysosomal storage disease caused by a deficient (or absent) enzyme, iduronate-2-sulfatase (I2S).[1][2] The accumulated substrate in Hunter’s syndrome is heparan sulfate and dermatan sulfate.[3] The syndrome has X-linked recessive inheritance.[3]
PathophysiologyHunter syndrome, or mucopolysaccharidosis II (MPS II), is a serious genetic disorder that primarily affects males (X-linked recessive). It interferes with the body’s ability to break down and recycle specific mucopolysaccharides, also known as glycosaminoglycans or GAG. Hunter syndrome is one of several related lysosomal storage diseases.
In Hunter syndrome, GAG builds up in cells throughout the body due to a deficiency or absence of the enzyme iduronate-2-sulfatase (I2S). This buildup interferes with the way certain cells and organs in the body function and leads to a number of serious symptoms. As the buildup of GAG continues throughout the cells of the body, signs of Hunter syndrome become more visible. Physical manifestations for some people with Hunter syndrome include distinct facial features and large head. In some cases of Hunter syndrome, central nervous system involvement leads to developmental delays and nervous system problems. Not all people with Hunter syndrome are affected by the disease in exactly the same way, and the rate of symptom progression varies widely. However, Hunter syndrome is always severe, progressive, and life-limiting.
Signs and symptomsThe symptoms of Hunter syndrome (MPS II) are generally not apparent at birth, but usually start to become noticeable after the first year of life. Often, the first symptoms of Hunter syndrome may include abdominal hernias, ear infections, runny noses, and colds. Since these symptoms are quite common among all infants, they are not likely to lead a doctor to make a diagnosis of Hunter syndrome right away. As the buildup of GAG continues throughout the cells of the body, signs of Hunter syndrome become more visible. Physical appearances of many children with Hunter syndrome include a distinctive coarseness in their facial features, including a prominent forehead, a nose with a flattened bridge, and an enlarged tongue. For this reason, unrelated children with Hunter syndrome often look alike. They may also have a large head as well as an enlarged abdomen. Many continue to have frequent infections of the ears and respiratory tract.
The continued storage of GAG in cells can lead to organs being affected in important ways. The thickening of the heart valves along with the walls of the heart can result in progressive decline in cardiac function. The walls of the airway may become thickened as well, leading to breathing problems while sleeping (obstructive airway disease). People with Hunter syndrome may also have limited lung capacity due to pulmonary involvement. As the liver and spleen grow larger with time, the belly may become distended, making hernias more noticeable. All major joints (including the wrists, elbows, shoulders, hips, and knees) may be affected by Hunter syndrome, leading to joint stiffness and limited motion. Progressive involvement of the finger and thumb joints results in decreased ability to pick up small objects. The effects on other joints, such as hips and knees, can make it increasingly difficult to walk normally. If carpal tunnel syndrome develops, a further decrease in hand function can occur. The bones themselves may be affected, resulting in short stature. In addition, pebbly, ivory-colored skin lesions** may be found on the **upper arms and legs and upper back of some people with Hunter syndrome. The presence or absence of the skin lesions is not helpful, however, in predicting clinical severity in Hunter syndrome. Finally, the storage of GAG in the brain can lead to delayed development with subsequent mental retardation. The rate and degree of progression may be different for each person with Hunter syndrome.
There is a broad range of severity in the symptoms of Hunter syndrome. It is important to note that though the term “mild” is used by physicians in comparing people with Hunter syndrome, the effects of even mild disease are quite serious. Two of the most significant areas of variability concern the degree of mental retardation and expected lifespan. Some people who have Hunter syndrome are not mentally retarded and live into their 20s or 30s; there are occasional reports of people who have lived into their 50s or 60s. The quality of life remains high in a large number of people, and many adults are actively employed.[citation needed] In contrast, others with Hunter syndrome develop severe mental impairment and have life expectancies of 15 years or fewer.

What is this syndrome which is histologically characterized by widely dispersed granular material amidst normal fibers?
1 Ehlers Danlos Syndrome
2 Pseudoxanthoma Elasticum
3 Buschke-Ollendorf Syndrome
4 Focal Dermal Hypoplasia
5 Lipoid Proteinosis

Pseudoxanthoma elasticum is genodermatosis characterized by redundant skin, angioid streaks, yellow papules on the mucous membranes and bleeding from gastric artery. On histology, readily apparent denerative changes of the elastic fibers are prominent, even without special stains.
Q/Q(M)-476806 Report a Problem
What is the most common genetic defect associated with this syndrome?
1 Neurofibromin
2 Merlin
3 Tuberin
4 Hamartin
5 Folliculin

Neurofibromatosis I occur due to a microdeletion at 17q11.2 involving the NF1 gene, which encodes for neurofibromin. It is an autosomal dominant disorder characterized by numerous benign tumors (neurofibromas) of the peripheral nervous system, café au lait macules, freckling in the area of the armpit (crow’s sign), two or more growths on the iris of the eye (known as Lisch nodules or iris hamartomas), tumor on the optic nerve (optic glioma), abnormal development of the spine (scoliosis), the temple (sphenoid) bone of the skull, or the tibia (one of the long bones of the shin) and a first degree relative (parent, sibling, or child) with NF1. The other proteins in the list are associated with other syndromes: In neurofibromatosis type 2, a NF2 gene mutation has been identified which encodes for a protein called Merlin, in tuberous sclerosis two genetic mutations have been identified on two separate chromosomes namely tuberin and hamartin, and a Folliculin mutation is seen in Birt Hogg Dube syndrome.
Q/Q(M)-482072 Report a Problem
What condition is associated with this finding of inflammatory keratotic facial papules which may result in scarring and atrophy?
1 Chloracne
2 Systemic lupus erythematosus
3 Keratosis pilaris
4 Reiter’s syndrome
5 Ulerythema ophryogenes

Ulerythema ophryogenes is a rare disorder that affects children and young adults. It is characterized by keratosis pilaris atrophicans and loss of lateral third of eyebrow.
Q/Q(M)-476831 Report a Problem
What is the genetic defect of this syndrome?
1 ABCA12 gene
2 Transglutaminase 1 (TGM1).
3 GJB2 (connexin 26) gene.
4 Steroid sulfatase (STS) gene
5 Profilaggrin gene

Mutations in the ABCA12 gene cause harlequin ichthyosis. X-linked ichthyosis (XLI) caused by a steroid sulfatase (STS) deficiency. Keratitis-ichthyosis-deafness syndrome is caused by a mutation in the GJB2 (connexin 26) gene. Ichthyosis vulgaris is caused by a defect in profilaggrin gene and lamellar ichthyosis involves a mutation in the gene for transglutaminase 1 (TGM1).
Q/Q(M)-482080 Report a Problem